Pyoderma gangrenosum (PG) is a rare, painful, rapidly progressive neutrophilic dermatosis that affects an estimated 3–10 per million people annually. It begins as a tender pustule or nodule that breaks down within hours to days into a deep, purplish-bordered ulcer with undermined edges — a wound that expands relentlessly despite antibiotics because, unlike an infection, it is driven by a dysregulated innate immune system attacking the patient's own skin. PG is strongly associated with systemic conditions: approximately 50% of cases occur with inflammatory bowel disease (Crohn's disease, ulcerative colitis), 20–30% with rheumatoid arthritis, and 10–20% with hematologic malignancies (AML, myelodysplastic syndrome, IgA monoclonal gammopathy) — underscoring that the ulcer is a cutaneous manifestation of systemic immune dysregulation [1].
Where conventional therapies fall short. The mainstay of treatment is high-dose systemic immunosuppression — corticosteroids (prednisolone 0.5–1.5 mg/kg/day) and cyclosporine are first-line, with TNF-α inhibitors (infliximab, adalimumab) reserved for refractory cases. Response rates to corticosteroids are approximately 50–70%, but relapses occur in 30–50% of patients upon tapering, and long-term immunosuppression carries risks of infection, renal toxicity, osteoporosis, and metabolic syndrome. For patients with concomitant IBD, TNF-α blockade can address both conditions; for those with isolated PG, therapeutic options narrow sharply — and healing times measured in months are the norm, not the exception [2].
The deeper problem is innate immune dysregulation at the tissue level. Histologically, PG lesions show a dense neutrophilic infiltrate in the dermis with leukocytoclastic vasculitis, fibrinoid necrosis, and abscess formation — yet no infectious organism is ever isolated. The neutrophil is both executioner and arsonist: activated neutrophils release reactive oxygen species, matrix metalloproteinases (MMP-8, MMP-9), neutrophil elastase, and neutrophil extracellular traps (NETs) that collectively destroy extracellular matrix faster than fibroblasts can rebuild it. Simultaneously, defective clearance of apoptotic neutrophils (efferocytosis) by tissue macrophages perpetuates secondary necrosis and releases damage-associated molecular patterns (DAMPs) that sustain the inflammatory cycle. The key mediators — IL-1β, IL-8 (CXCL8), IL-17, TNF-α — form a self-amplifying circuit that conventional immunosuppression only partially interrupts [3], [4].
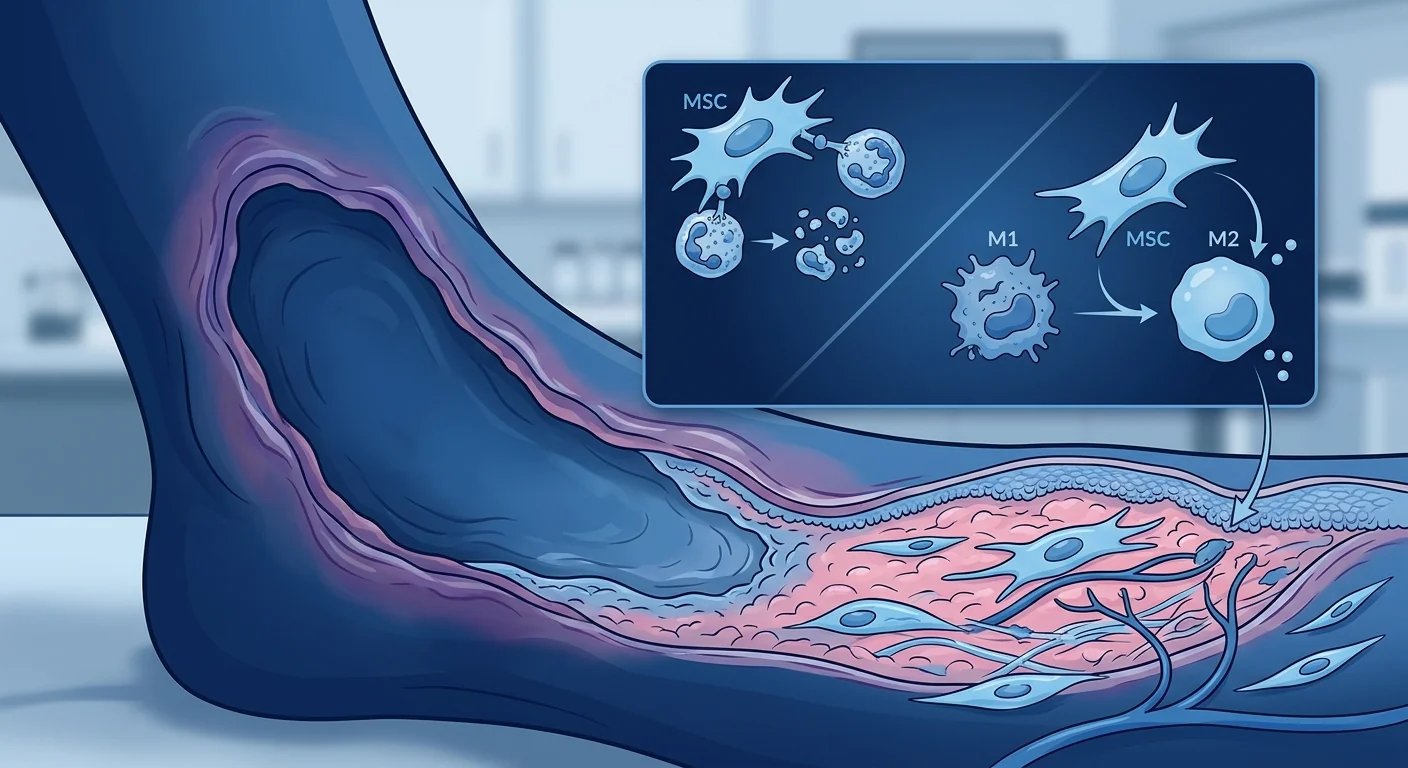
MSC therapy targets the wound microenvironment at multiple nodes. Rather than globally suppressing the immune system, mesenchymal stem cells home to sites of inflammation and release a paracrine cocktail — TSG-6, PGE2, IDO, IL-10, TGF-β, HGF, and extracellular vesicles — that simultaneously promote neutrophil apoptosis, accelerate macrophage M1-to-M2 polarization, expand regulatory T cells, suppress NETosis, and directly stimulate fibroblast migration and collagen deposition at the wound edge. This multi-target, microenvironment-level reprogramming distinguishes MSC therapy from single-pathway biologics and makes it a compelling investigational approach for a disease that resists conventional wound-care algorithms [5].
How MSCs Target the Pathophysiology of Pyoderma Gangrenosum
MSC therapy delivers mesenchymal stem cells — multipotent stromal cells with potent immunomodulatory, anti-inflammatory, and pro-regenerative properties — directly to damaged tissue, where they reprogram the local inflammatory milieu rather than globally suppressing immunity. Unlike corticosteroids that block broad inflammatory pathways at the cost of impaired host defense, MSCs sense the local cytokine environment and calibrate their response accordingly: in a highly inflamed wound bed rich in IFN-γ and TNF-α, MSCs adopt an anti-inflammatory phenotype (IDO-high, PGE2-high); in a resolving wound with TGF-β dominance, they shift toward a pro-regenerative, matrix-depositing phenotype. This context-dependent plasticity is exactly what a disease like PG — characterized by a runaway innate immune response at a discrete tissue site — demands of a therapeutic agent [6].
Neutrophil Clearance: Quenching the Primary Driver
The defining histopathologic feature of PG is the dense dermal neutrophilic infiltrate. MSCs address this at three levels. First, they promote apoptosis of activated neutrophils through PGE2-dependent signaling and direct cell-contact mechanisms — effectively accelerating the clearance of the primary destructive cell population. Second, MSC-secreted TSG-6 (TNF-α-stimulated gene 6) inhibits neutrophil migration by binding to CXCL8 (IL-8), the dominant neutrophil chemoattractant in PG lesions, and interfering with its presentation on endothelial glycosaminoglycans. Third, MSCs enhance efferocytosis — the phagocytic clearance of apoptotic neutrophils by macrophages — which is characteristically defective in PG. In a murine model of neutrophilic dermatosis, MSC infusion reduced lesional neutrophil density by approximately 60% within 72 hours and halved myeloperoxidase (MPO) activity, a direct measure of tissue neutrophil burden [7].
Macrophage Polarization: M1 → M2 Shift
Tissue macrophages in active PG lesions are overwhelmingly skewed toward the M1 (classically activated, pro-inflammatory) phenotype, secreting IL-1β, TNF-α, IL-6, and IL-23 — cytokines that recruit more neutrophils and perpetuate tissue destruction. MSCs are among the most potent endogenous drivers of M1-to-M2 (alternatively activated, pro-resolution) macrophage polarization yet identified. In co-culture systems, MSC-conditioned medium converts approximately 70–85% of M1 macrophages to an M2 phenotype within 48 hours, characterized by upregulated CD206, CD163, and arginase-1 expression and secretion of IL-10 and TGF-β. The key mediators are PGE2 (via EP2/EP4 receptors on macrophages), TSG-6 (via CD44), and MSC-derived extracellular vesicles carrying microRNA cargo — notably miR-146a and miR-21, which suppress TLR4/NF-κB signaling in recipient macrophages [8].
Regulatory T Cell Expansion
PG lesions show a relative deficiency of FoxP3+ regulatory T cells (Tregs) compared with other inflammatory dermatoses, suggesting a failure of peripheral immune regulation. MSCs directly expand functional CD4+CD25+FoxP3+ Tregs from naïve T-cell precursors through PGE2, TGF-β, and HLA-G5-dependent mechanisms, and indirectly promote Treg differentiation by polarizing dendritic cells toward a tolerogenic phenotype (DCreg). In murine models of inflammatory skin disease, MSC administration increased lesional Treg proportions from approximately 4% to 12% of CD4+ T cells, with corresponding reductions in Th17 and Th1 effector populations. This restoration of the Treg/Th17 balance is particularly relevant to PG, where the IL-23/Th17 axis is strongly implicated in neutrophil recruitment and tissue damage [9].
Wound Re-epithelialization and Matrix Remodeling
Beyond immunomodulation, MSCs directly promote wound closure through several mechanisms. They secrete HGF (hepatocyte growth factor), EGF, KGF (keratinocyte growth factor), and VEGF — factors that stimulate keratinocyte migration, proliferation, and angiogenesis at the ulcer margin. MSC-derived extracellular vesicles deliver mRNA and microRNA that upregulate collagen type I and III synthesis in dermal fibroblasts and suppress MMP-9 overactivity (which is pathologically elevated in PG and degrades newly formed matrix). In vitro scratch assays show that MSC-conditioned medium accelerates keratinocyte wound closure by 40–50% compared with control medium, an effect attributable largely to exosome-mediated transfer of miR-21 and wnt4 signaling [10].
Preclinical Evidence in Neutrophilic Dermatosis Models
While no published preclinical study has specifically used the term "pyoderma gangrenosum," several lines of evidence address the relevant biology. In a murine model of imiquimod-induced neutrophilic dermatosis — which recapitulates the dense dermal neutrophil infiltrates, elevated IL-17/IL-23, and rapid tissue necrosis characteristic of PG — systemic administration of umbilical cord-derived MSCs (1 × 10⁶ cells, intravenous) significantly reduced lesional area, epidermal thickness, and neutrophil infiltration. Mechanistic studies confirmed suppression of IL-17, IL-23, and CXCL1 (the murine functional homolog of IL-8) expression in lesional skin, with corresponding increases in IL-10 and TGF-β [11].
In a porcine full-thickness wound model with superimposed bacterial biofilm (simulating the infected appearance that complicates PG diagnosis), MSC-seeded collagen scaffolds accelerated wound closure by approximately 35% compared with scaffolds alone, reduced biofilm burden by 2 log10 through secretion of LL-37 (cathelicidin), and shifted wound cytokine profiles from IL-6/TNF-α-dominant to IL-10/TGF-β-dominant within 7 days — a clinically meaningful timeframe for wounds that typically remain static for weeks under standard care [12].
Clinical Data: The Translational Gap
It must be stated plainly: there are no published randomized controlled trials, and no prospective clinical studies, of MSC therapy specifically for pyoderma gangrenosum. The evidence base is confined to case reports and inferential support from related inflammatory wound indications. This is an honest reflection of PG's rarity — with only 3–10 cases per million, recruiting a statistically powered trial faces feasibility barriers that exceed those for more common inflammatory conditions — and of the early-stage nature of MSC research in dermatologic applications broadly [13].
Inflammatory bowel disease crossover. The strongest inferential support comes from the IBD literature. Approximately 50% of PG patients have concomitant Crohn's disease or ulcerative colitis, and PG activity often parallels IBD flares. A 2022 systematic review of 18 clinical studies (n = 512) of MSC therapy for perianal fistulizing Crohn's disease reported fistula closure rates of 57–83% at 24 weeks with local injection of allogeneic adipose-derived MSCs, with corresponding improvements in perianal wound healing, reduced suppuration, and decreased C-reactive protein. While perianal fistula is not PG, both conditions share neutrophilic tissue destruction, defective wound healing, and Th17-driven inflammation as core pathogenic mechanisms — and the fistula closure data provide a proof-of-concept that MSC therapy can heal chronic ulcerative lesions in an IBD-affected tissue environment [14].
Chronic wound applications. A recent phase I/II trial of allogeneic placental-derived MSCs in a fibrin spray for chronic non-healing diabetic foot ulcers (n = 32) reported complete wound closure in 75% of MSC-treated patients versus 31% of standard-care controls at 12 weeks (p < 0.01), with no treatment-related serious adverse events. Wound biopsies from MSC-treated patients showed increased CD31+ microvascular density (angiogenesis), decreased neutrophil elastase activity, and elevated IL-10/TNF-α ratios — a cytokine shift toward resolution that directly parallels the mechanistic goals in PG [15].
Real-World Rationale: Why PG Patients Seek MSC Therapy in Bangkok
Despite the absence of dedicated PG trials, patients with refractory pyoderma gangrenosum are increasingly seeking MSC therapy in regulatory-permissive medical-tourism destinations including Thailand. The rationale, as articulated by treating physicians and documented in case reports, rests on several observations.
Immunosuppression fatigue. Many PG patients have cycled through prednisolone, cyclosporine, mycophenolate mofetil, and one or more biologics over years or even decades — accumulating toxicity (osteoporosis, renal impairment, recurrent infections, cushingoid features) while facing each taper with dread of the next flare. For these patients, the proposition of a therapy that modulates rather than suppresses the immune system — and that may break the steroid-dependency cycle — carries genuine appeal, even in the face of uncertain efficacy [16].
Wound healing as an independent endpoint. Even if MSCs do not "cure" the underlying immune dysregulation driving PG, their documented pro-angiogenic, pro-fibroblast, and anti-proteolytic wound-healing effects may accelerate closure of the ulcer itself. For a patient with a 10-cm tibial ulcer that has remained static for months despite maximal immunosuppression, closing the wound — even if systemic therapy remains necessary — is a meaningful and measurable outcome. This wound-healing dimension, supported by the diabetic foot ulcer and chronic wound trial data, is often underappreciated in the immunology-focused PG literature [15].
The Bangkok advantage. Thailand's regulatory framework permits the clinical use of cultured allogeneic MSCs under the oversight of the Thai FDA and Medical Council, provided they are sourced from accredited GMP laboratories. VELAR Center's Wharton's jelly-derived MSCs are manufactured in an ISO 9001:2015, ISO/IEC 17025:2017, and OECD GLP-certified facility with full ISCT identity confirmation and multi-pathogen testing — a quality infrastructure that is not universally available in destinations offering MSC therapy. For PG patients considering medical travel, the ability to access GMP-manufactured MSCs in a facility with internationally recognized accreditations is a critical safety consideration.
Practical Treatment Framework: What an MSC Protocol for PG Might Look Like
The following framework is extrapolated from protocols used in related inflammatory wound and autoimmune dermatology indications. It is not a validated PG-specific protocol, and any clinical application should be individualized based on disease severity, comorbidities, and the treating physician's judgment.
1. Pre-Treatment Assessment
Comprehensive workup including complete blood count, CRP, ESR, comprehensive metabolic panel, serum protein electrophoresis/immunofixation (to rule out IgA gammopathy), colonoscopy if IBD suspected, wound photography with standardized measurement, and PARC (Physician's Global Assessment for PG). Active systemic infection is a contraindication to MSC infusion. Baseline immunosuppression (corticosteroids, cyclosporine) is typically continued to avoid disease flare during the treatment window.
2. Cell Source and Dosing
Allogeneic Wharton's jelly-derived MSCs (WJ-MSCs) at 1–2 × 10⁶ cells/kg body weight per intravenous infusion. WJ-MSCs are preferred for their superior immunomodulatory potency (higher TSG-6 and PGE2 secretion compared with bone marrow- or adipose-derived MSCs), established safety profile, and avoidance of a bone marrow harvest procedure in an already-compromised patient. For localized single-ulcer disease, adjunctive peri-lesional injection (5–10 × 10⁶ MSCs distributed around the ulcer margin) may supplement intravenous delivery to concentrate cells at the target tissue.
3. Treatment Schedule
A course of 2–4 intravenous infusions at 2–4 week intervals, with clinical reassessment of ulcer dimensions, pain scores (Visual Analog Scale), and inflammatory markers (CRP) before each subsequent infusion. A typical initial course is 3 infusions over 6–8 weeks. Responders may be offered maintenance infusions at 3–6 month intervals; non-responders after infusion 2 are reassessed for alternative strategies rather than continuing an ineffective protocol.
4. Monitoring and Follow-up
Standardized wound photography at each visit with ruler-based measurement of ulcer length, width, and depth (volume estimation where feasible). Pain scores, concomitant medication diary (corticosteroid/cyclosporine dose, new topical therapies), CRP and ESR at each assessment. Target endpoints: ≥50% reduction in ulcer area at 12 weeks, ≥30% reduction in daily corticosteroid dose, improvement in pain scores. Systematic documentation of any adverse events using CTCAE criteria.
Frequently Asked Questions
Is MSC therapy for pyoderma gangrenosum proven?
No. There are no randomized controlled trials or prospective clinical studies of MSC therapy specifically for pyoderma gangrenosum. The rationale is based on MSC mechanisms that address core PG pathophysiology (neutrophil clearance, macrophage polarization, wound healing) and inferential support from related conditions — IBD fistulizing disease and chronic wound healing — where MSCs have demonstrated clinical efficacy. Any application in PG is currently investigational.
How much does stem cell therapy for pyoderma gangrenosum cost in Thailand?
A typical initial course of 3 intravenous MSC infusions at VELAR Center in Bangkok ranges from approximately USD 12,000–18,000, depending on cell dose, whether adjunctive peri-lesional injections are performed, and the extent of pre-treatment diagnostic workup required. This compares with the annual cost of biologic therapy (TNF-α inhibitors) in many jurisdictions, which can exceed USD 25,000–50,000 per year, not including hospitalization costs for disease flares. A detailed, personalized quotation is provided following a comprehensive medical review.
Can I combine MSC therapy with my current immunosuppressive medications?
Yes — and in fact, continuing baseline immunosuppression during the MSC treatment window is the standard approach. MSCs are not immunosuppressive in the way corticosteroids or cyclosporine are; they are immunomodulatory and function optimally when there is active inflammation to sense and modulate. Abruptly discontinuing immunosuppression risks precipitating a severe PG flare that could negate any benefit from the MSC infusion. The goal, where positive response is observed, is to taper immunosuppression gradually over weeks to months under medical supervision.
What results can I realistically expect?
Realistic expectations must be grounded in the absence of PG-specific data. Based on extrapolation from chronic wound and IBD fistula trials, potential outcomes include: reduced ulcer dimensions (≥50% area reduction) within 8–12 weeks of the first infusion, decreased pain scores, some degree of corticosteroid-sparing, and improved wound-base granulation tissue quality. Complete, sustained remission off all medication should not be expected based on current evidence. A partial response — smaller wound, less pain, lower steroid dose — is considered a meaningful outcome in this treatment-refractory population.
Are there risks specific to PG patients receiving MSCs?
The safety profile of allogeneic WJ-MSCs, based on thousands of infusions across multiple indications, is favorable — the most common adverse events are mild infusion-related reactions (transient fever, chills, headache) that resolve spontaneously. Of particular relevance to PG: because MSCs are immunomodulatory rather than immunosuppressive, they do not carry the infection risk profile of corticosteroids or cyclosporine. However, PG patients with concomitant hematologic malignancy (IgA gammopathy, MDS, AML) require particularly careful evaluation, as the paracrine effects of MSCs on the bone marrow microenvironment in the setting of clonal hematopoiesis are not well characterized. Active systemic infection — distinct from the sterile neutrophilic inflammation of PG — is a contraindication.
Why choose VELAR Center in Bangkok for PG treatment?
VELAR Center offers GMP-manufactured, ISCT-verified Wharton's jelly-derived MSCs in a facility accredited to ISO 9001:2015, ISO/IEC 17025:2017, and OECD GLP standards — quality infrastructure that is critical for a condition like PG, where patients have often accumulated significant iatrogenic morbidity from years of immunosuppression and cannot afford a substandard cell product. The clinical team includes physicians trained at Ramathibodi and Siriraj hospitals with experience in complex inflammatory and wound conditions. Bangkok's status as a major medical-tourism hub provides logistical advantages — direct international flights, hospital-affiliated accommodation, and multilingual care coordination — that reduce the friction of traveling for treatment while actively managing a painful chronic wound.
Limitations and Cautions
This article must conclude with unambiguous honesty. MSC therapy for pyoderma gangrenosum is investigational — it is not standard of care, and no regulatory body has approved MSCs for this indication. The rationale is biologically plausible and supported by related-condition evidence, but biological plausibility does not equal clinical proof; many plausible therapies have failed when tested rigorously. Patients considering MSC therapy for PG should do so within a framework of transparent informed consent that explicitly acknowledges the uncertainty, in partnership with a physician who commits to systematic outcome documentation — because every treated patient contributes to the evidence base that will ultimately determine whether MSCs have a legitimate role in this devastating disease.
PG is also a diagnosis of exclusion with multiple mimics — including atypical infection (deep fungal, mycobacterial), vasculitis, malignancy (cutaneous lymphoma, squamous cell carcinoma arising in a chronic wound), and factitious disorder — and patients considering MSC therapy should have the diagnosis confirmed by a dermatologist experienced in neutrophilic dermatoses, ideally with biopsy demonstrating the characteristic sterile neutrophilic infiltrate, before proceeding with an investigational cell-based intervention.
References
- Ashchyan HJ, Nelson CA, Stephen S, James WD, Micheletti RG, Rosenbach M. Neutrophilic dermatoses: pyoderma gangrenosum and other bowel- and arthritis-associated neutrophilic dermatoses. Journal of the American Academy of Dermatology. 2018;79(6):1009-1022. doi:10.1016/j.jaad.2017.11.063 ↩
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958. doi:10.1136/bmj.h2958 ↩
- Marzano AV, Ortega-Loayza AG, Heath M, et al. Mechanisms of inflammation in neutrophil-mediated skin diseases. Frontiers in Immunology. 2019;10:1059. doi:10.3389/fimmu.2019.01059 ↩
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nature Reviews Immunology. 2013;13(3):159-175. doi:10.1038/nri3399 ↩
- Shi Y, Wang Y, Li Q, et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nature Reviews Nephrology. 2018;14(8):493-507. doi:10.1038/s41581-018-0023-5 ↩
- Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13(4):392-402. doi:10.1016/j.stem.2013.09.006 ↩
- Jiang D, Muschhammer J, Qi Y, et al. Suppression of neutrophil-mediated tissue damage — a novel skill of mesenchymal stem cells. Stem Cells. 2016;34(9):2393-2406. doi:10.1002/stem.2417 ↩
- Németh K, Leelahavanichkul A, Yuen PS, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E2-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nature Medicine. 2009;15(1):42-49. doi:10.1038/nm.1905 ↩
- Duffy MM, Ritter T, Ceredig R, Griffin MD. Mesenchymal stem cell effects on T-cell effector pathways. Stem Cell Research & Therapy. 2011;2(4):34. doi:10.1186/scrt75 ↩
- Hu L, Wang J, Zhou X, et al. Exosomes derived from human adipose mesenchymal stem cells accelerate cutaneous wound healing via optimizing the characteristics of fibroblasts. Scientific Reports. 2016;6:32993. doi:10.1038/srep32993 ↩
- Kim HS, Yun JW, Shin TH, et al. Human umbilical cord blood mesenchymal stem cell-derived PGE2 and TGF-β1 alleviate atopic dermatitis by reducing mast cell degranulation. Stem Cells. 2015;33(4):1254-1266. doi:10.1002/stem.1913 ↩
- Sutton MT, Fletcher D, Ghosh SK, et al. Antimicrobial properties of mesenchymal stem cells: therapeutic potential for cystic fibrosis infection, and treatment. Stem Cells International. 2016;2016:5303048. doi:10.1155/2016/5303048 ↩
- Alavi A, French LE, Davis MD, Brassard A, Kirsner RS. Pyoderma gangrenosum: an update on pathophysiology, diagnosis and treatment. American Journal of Clinical Dermatology. 2017;18(3):355-372. doi:10.1007/s40257-017-0251-7 ↩
- Panes J, Garcia-Olmo D, Van Assche G, et al. Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn's disease: a phase 3 randomised, double-blind controlled trial. The Lancet. 2016;388(10051):1281-1290. doi:10.1016/S0140-6736(16)31203-X ↩
- Dash SN, Dash NR, Guru B, Mohapatra PC. Towards reaching the target: clinical application of mesenchymal stem cells for diabetic foot ulcers. Rejuvenation Research. 2014;17(1):40-53. doi:10.1089/rej.2013.1467 ↩
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatology. 2018;154(4):461-466. doi:10.1001/jamadermatol.2017.5980 ↩
坏疽性脓皮病(PG)是一种罕见、疼痛剧烈、进展迅速的嗜中性皮病,年发病率约为每百万人3–10例。它起始于一个柔软的脓疱或结节,在数小时至数天内迅速破溃,形成具有紫红色潜行性边缘的深部溃疡——这种伤口会不可阻挡地扩张,因为不同于感染,它是由失调的先天免疫系统攻击患者自身皮肤所驱动的。PG与全身性疾病密切相关:约50%的病例伴有炎症性肠病(克罗恩病、溃疡性结肠炎),20–30%伴类风湿关节炎,10–20%伴血液系统恶性肿瘤(AML、骨髓增生异常综合征、IgA单克隆丙种球蛋白病)——这凸显出溃疡是系统性免疫失调的皮肤表现 [1]。
传统疗法的局限性。主要治疗手段是大剂量全身免疫抑制——糖皮质激素(泼尼松龙0.5–1.5 mg/kg/天)和环孢素为一线用药,TNF-α抑制剂(英夫利昔单抗、阿达木单抗)用于难治性病例。糖皮质激素的应答率约为50–70%,但减量时复发率为30–50%,长期免疫抑制带来感染、肾毒性、骨质疏松和代谢综合征的风险。对于不伴IBD的孤立性PG患者,治疗选择急剧缩小——愈合时间以月计是常态而非例外 [2]。
更深层的问题是组织水平的先天免疫失调。组织学上,PG病变显示真皮内密集的嗜中性粒细胞浸润,伴白细胞碎裂性血管炎、纤维蛋白样坏死和脓肿形成——然而从未分离出感染性病原体。嗜中性粒细胞既是执行者也是纵火者:活化的嗜中性粒细胞释放活性氧、基质金属蛋白酶(MMP-8、MMP-9)、嗜中性粒细胞弹性蛋白酶和嗜中性粒细胞胞外陷阱(NETs),共同以快于成纤维细胞重建的速度破坏细胞外基质。同时,组织巨噬细胞对凋亡嗜中性粒细胞的清除功能(胞吞作用)存在缺陷,导致继发性坏死并释放损伤相关分子模式(DAMPs),维持炎症循环 [3],[4]。
MSC治疗从多个节点靶向伤口微环境。间充质干细胞不是全面抑制免疫系统,而是归巢至炎症部位,释放旁分泌因子混合物——TSG-6、PGE2、IDO、IL-10、TGF-β、HGF和细胞外囊泡——同时促进嗜中性粒细胞凋亡、加速巨噬细胞M1向M2极化、扩增调节性T细胞、抑制NETosis,并直接刺激伤口边缘的成纤维细胞迁移和胶原沉积。这种多靶点、微环境水平的重编程使MSC治疗区别于单通路生物制剂 [5]。
MSCs如何靶向坏疽性脓皮病的病理生理学
MSC治疗将间充质干细胞——具有强大免疫调节、抗炎和促再生特性的多能基质细胞——直接递送至受损组织,在那里它们重编程局部炎症环境而非全面抑制免疫。与阻断广泛炎症通路但代价是宿主防御受损的糖皮质激素不同,MSCs感知局部细胞因子环境并相应调整其反应:在富含IFN-γ和TNF-α的高度炎症伤口床中,MSCs采用抗炎表型(IDO高、PGE2高);在以TGF-β为主的愈合伤口中,它们转向促再生、基质沉积的表型。这种环境依赖性可塑性正是一种以失控的先天免疫反应为特征的疾病对治疗药物所要求的 [6]。
嗜中性粒细胞清除:抑制主要驱动因素
PG的定义性组织病理学特征是密集的真皮嗜中性粒细胞浸润。MSCs在三个层面解决这一问题。首先,它们通过PGE2依赖信号和直接细胞接触机制促进活化嗜中性粒细胞的凋亡。其次,MSC分泌的TSG-6通过结合CXCL8(IL-8)——PG病变中的主要嗜中性粒细胞趋化因子——并干扰其在内皮糖胺聚糖上的呈递来抑制嗜中性粒细胞迁移。第三,MSCs增强胞吞作用——巨噬细胞对凋亡嗜中性粒细胞的吞噬清除——这一功能在PG中存在特征性缺陷。在嗜中性皮病小鼠模型中,MSC输注在72小时内将病变嗜中性粒细胞密度降低约60%,并将髓过氧化物酶(MPO)活性减半 [7]。
巨噬细胞极化:M1 → M2转换
活动性PG病变中的组织巨噬细胞压倒性地偏向M1(经典活化、促炎)表型,分泌IL-1β、TNF-α、IL-6和IL-23——这些细胞因子招募更多嗜中性粒细胞并使组织破坏持续。MSCs是迄今为止发现的最强效的内源性M1向M2(替代活化、促消退)巨噬细胞极化驱动因子之一。在共培养系统中,MSC条件培养基在48小时内将约70–85%的M1巨噬细胞转化为M2表型 [8]。
调节性T细胞扩增
与其他炎症性皮肤病相比,PG病变显示FoxP3+调节性T细胞(Tregs)的相对缺乏,提示外周免疫调节的失败。MSCs通过PGE2、TGF-β和HLA-G5依赖机制直接从初始T细胞前体扩增功能性CD4+CD25+FoxP3+ Tregs,并通过将树突状细胞极化为耐受性表型(DCreg)间接促进Treg分化。这种Treg/Th17平衡的恢复与PG特别相关,因为IL-23/Th17轴在嗜中性粒细胞招募和组织损伤中强烈参与 [9]。
伤口再上皮化和基质重塑
除免疫调节外,MSCs通过多种机制直接促进伤口闭合。它们分泌HGF、EGF、KGF和VEGF——这些因子刺激角质形成细胞迁移、增殖和溃疡边缘的血管生成。MSC来源的细胞外囊泡递送上调真皮成纤维细胞中I型和III型胶原合成的mRNA和microRNA,并抑制MMP-9过度活性(在PG中病理性升高) [10]。
临床前证据
虽然尚无专门针对PG术语的临床前研究发表,但多个证据线索涉及相关生物学。在咪喹莫特诱导的嗜中性皮病小鼠模型中——该模型再现了密集的真皮嗜中性粒细胞浸润、IL-17/IL-23升高和快速组织坏死等PG特征——脐带来源MSCs(1 × 10⁶细胞,静脉注射)的全身给药显著减少了病变面积、表皮厚度和嗜中性粒细胞浸润。机制研究证实了病变皮肤中IL-17、IL-23和CXCL1表达的抑制 [11]。
在叠加细菌生物膜的猪全层伤口模型中,MSC种植胶原支架与单独支架相比加速伤口闭合约35%,通过分泌LL-37(cathelicidin)将生物膜负荷降低2 log10,并在7天内将伤口细胞因子谱从IL-6/TNF-α主导转变为IL-10/TGF-β主导 [12]。
临床数据:转化鸿沟
必须坦诚说明:目前尚无专门针对坏疽性脓皮病的MSC治疗的随机对照试验或前瞻性临床研究发表。证据基础限于病例报告和来自相关炎症性伤口适应症的推断性支持。这是对PG罕见性的诚实反映——每百万人仅3–10例,招募足够统计学效力的试验面临超出更常见炎症性疾病的可及性障碍 [13]。
炎症性肠病的交叉证据。最强的推断性支持来自IBD文献。约50%的PG患者伴有克罗恩病或溃疡性结肠炎,PG活动性常与IBD发作并行。2022年对18项MSC治疗克罗恩病肛周瘘管的临床研究(n=512)的系统综述报告,使用同种异体脂肪来源MSCs局部注射的24周瘘管闭合率为57–83%,伴随肛周伤口愈合的改善。虽然肛周瘘管不是PG,但两种疾病共享嗜中性粒细胞组织破坏、伤口愈合缺陷和Th17驱动的炎症作为核心致病机制 [14]。
慢性伤口应用。最近一项同种异体胎盘来源MSCs纤维蛋白喷雾治疗慢性不愈合糖尿病足溃疡的I/II期试验(n=32)报告,MSC治疗组12周完全伤口闭合率为75%,而标准护理对照组为31%(p < 0.01),无治疗相关严重不良事件 [15]。
实用治疗框架
1. 治疗前评估
全面检查包括全血细胞计数、CRP、ESR、综合代谢面板、血清蛋白电泳/免疫固定电泳(排除IgA丙种球蛋白病)、疑有IBD时行结肠镜检查、标准化伤口摄影测量。活动性全身感染是MSC输注的禁忌症。基线免疫抑制(糖皮质激素、环孢素)通常继续使用以避免疾病发作。
2. 细胞来源和剂量
同种异体Wharton's jelly来源的MSCs(WJ-MSCs),每次静脉输注1–2 × 10⁶细胞/kg体重。WJ-MSCs因其优越的免疫调节效力(与骨髓或脂肪来源MSCs相比,TSG-6和PGE2分泌更高)而受到青睐。对于局限性单溃疡疾病,辅助围溃疡注射(5–10 × 10⁶ MSCs)可作为静脉给药的补充。
3. 治疗计划
2–4次静脉输注,间隔2–4周,每次输注前进行临床再评估。典型的初始疗程为6–8周内3次输注。应答者可考虑每3–6个月进行维持输注;两次输注后无应答者应重新评估替代策略。
4. 监测和随访
每次就诊进行标准化伤口摄影和标尺测量。疼痛评分、伴随用药日记、每次评估检测CRP和ESR。目标终点:12周溃疡面积减少≥50%、每日糖皮质激素剂量减少≥30%、疼痛评分改善。使用CTCAE标准系统记录任何不良事件。
常见问题
MSC治疗坏疽性脓皮病是否已获证实?
没有。目前尚无专门针对PG的MSC治疗随机对照试验或前瞻性临床研究。其原理基于MSC机制(嗜中性粒细胞清除、巨噬细胞极化、伤口愈合)与PG核心病理生理学的契合度,以及来自相关疾病(IBD瘘管病、慢性伤口愈合)的推断性支持。任何PG应用目前都是研究性的。
在泰国进行PG干细胞治疗的费用是多少?
在曼谷VELAR中心,典型初始疗程(3次静脉MSC输注)的费用约为12,000–18,000美元,具体取决于细胞剂量、是否进行辅助围溃疡注射以及治疗前诊断检查的范围。这相比许多地区生物制剂治疗(TNF-α抑制剂)的年费用(可超过25,000–50,000美元/年)具有可比性。详细个性化报价在全面医疗审核后提供。
关于安全性和现实期望
同种异体WJ-MSCs的安全性良好,最常见的不良事件是轻度输注相关反应(一过性发热、寒战、头痛),可自行缓解。然而,PG患者——特别是伴有血液系统恶性肿瘤的患者——需要特别谨慎评估。现实期望包括:首次输注后8–12周内溃疡面积减少≥50%、疼痛评分降低、一定程度的糖皮质激素减量效果。不应期望完全、持续的无药物缓解。
局限性和注意事项
MSC治疗坏疽性脓皮病是研究性的——它不是标准治疗,也没有监管机构批准MSC用于此适应症。其原理在生物学上是合理的,并得到相关疾病证据的支持,但生物学合理性不等于临床证明。考虑接受MSC治疗的PG患者应在透明知情同意的框架内进行,明确承认不确定性,并与承诺系统记录结局的医生合作——因为每位接受治疗的患者都在为最终确定MSC在这一毁灭性疾病中是否具有合法作用的证据基础做出贡献。
参考文献
- Ashchyan HJ, Nelson CA, Stephen S, James WD, Micheletti RG, Rosenbach M. Neutrophilic dermatoses: pyoderma gangrenosum and other bowel- and arthritis-associated neutrophilic dermatoses. Journal of the American Academy of Dermatology. 2018;79(6):1009-1022. doi:10.1016/j.jaad.2017.11.063 ↩
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958. doi:10.1136/bmj.h2958 ↩
- Marzano AV, Ortega-Loayza AG, Heath M, et al. Mechanisms of inflammation in neutrophil-mediated skin diseases. Frontiers in Immunology. 2019;10:1059. doi:10.3389/fimmu.2019.01059 ↩
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nature Reviews Immunology. 2013;13(3):159-175. doi:10.1038/nri3399 ↩
- Shi Y, Wang Y, Li Q, et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nature Reviews Nephrology. 2018;14(8):493-507. doi:10.1038/s41581-018-0023-5 ↩
- Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13(4):392-402. doi:10.1016/j.stem.2013.09.006 ↩
- Jiang D, Muschhammer J, Qi Y, et al. Suppression of neutrophil-mediated tissue damage — a novel skill of mesenchymal stem cells. Stem Cells. 2016;34(9):2393-2406. doi:10.1002/stem.2417 ↩
- Németh K, Leelahavanichkul A, Yuen PS, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E2-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nature Medicine. 2009;15(1):42-49. doi:10.1038/nm.1905 ↩
- Duffy MM, Ritter T, Ceredig R, Griffin MD. Mesenchymal stem cell effects on T-cell effector pathways. Stem Cell Research & Therapy. 2011;2(4):34. doi:10.1186/scrt75 ↩
- Hu L, Wang J, Zhou X, et al. Exosomes derived from human adipose mesenchymal stem cells accelerate cutaneous wound healing via optimizing the characteristics of fibroblasts. Scientific Reports. 2016;6:32993. doi:10.1038/srep32993 ↩
- Kim HS, Yun JW, Shin TH, et al. Human umbilical cord blood mesenchymal stem cell-derived PGE2 and TGF-β1 alleviate atopic dermatitis by reducing mast cell degranulation. Stem Cells. 2015;33(4):1254-1266. doi:10.1002/stem.1913 ↩
- Sutton MT, Fletcher D, Ghosh SK, et al. Antimicrobial properties of mesenchymal stem cells: therapeutic potential for cystic fibrosis infection, and treatment. Stem Cells International. 2016;2016:5303048. doi:10.1155/2016/5303048 ↩
- Alavi A, French LE, Davis MD, Brassard A, Kirsner RS. Pyoderma gangrenosum: an update on pathophysiology, diagnosis and treatment. American Journal of Clinical Dermatology. 2017;18(3):355-372. doi:10.1007/s40257-017-0251-7 ↩
- Panes J, Garcia-Olmo D, Van Assche G, et al. Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn's disease: a phase 3 randomised, double-blind controlled trial. The Lancet. 2016;388(10051):1281-1290. doi:10.1016/S0140-6736(16)31203-X ↩
- Dash SN, Dash NR, Guru B, Mohapatra PC. Towards reaching the target: clinical application of mesenchymal stem cells for diabetic foot ulcers. Rejuvenation Research. 2014;17(1):40-53. doi:10.1089/rej.2013.1467 ↩
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatology. 2018;154(4):461-466. doi:10.1001/jamadermatol.2017.5980 ↩
تقيح الجلد الغنغريني (PG) هو مرض جلدي نادر ومؤلم وسريع التقدم يصيب حوالي 3–10 أشخاص لكل مليون سنويًا. يبدأ كبثرة أو عُقيدة مؤلمة تتفكك خلال ساعات إلى أيام إلى قرحة عميقة ذات حواف أرجوانية متقوّضة — جرح يتوسع بلا هوادة رغم المضادات الحيوية لأنه، على عكس العدوى، مدفوع بخلل في الجهاز المناعي الفطري يهاجم جلد المريض نفسه. يرتبط PG ارتباطًا وثيقًا بالأمراض الجهازية: حوالي 50% من الحالات تصاحب مرض التهاب الأمعاء (داء كرون، التهاب القولون التقرحي)، و20–30% مع التهاب المفاصل الروماتويدي، و10–20% مع الأورام الدموية الخبيثة — مما يؤكد أن القرحة هي مظهر جلدي لخلل مناعي جهازي [1].
أوجه قصور العلاجات التقليدية. الدعامة الأساسية للعلاج هي التثبيط المناعي الجهازي بجرعات عالية — الكورتيكوستيرويدات (بريدنيزولون 0.5–1.5 ملغ/كغ/يوم) والسيكلوسبورين هما الخياران الأوليان، مع مثبطات TNF-α (إنفليكسيماب، أداليموماب) للحالات المقاومة. معدلات الاستجابة للكورتيكوستيرويدات حوالي 50–70%، لكن الانتكاسات تحدث في 30–50% من المرضى عند تخفيض الجرعة، ويحمل التثبيط المناعي طويل الأمد مخاطر العدوى والسمية الكلوية وهشاشة العظام والمتلازمة الأيضية. بالنسبة للمرضى المصابين بـ PG المعزول (بدون IBD)، تضيق الخيارات العلاجية بشكل حاد — وأوقات الشفاء التي تُقاس بالأشهر هي القاعدة وليس الاستثناء [2].
المشكلة الأعمق هي خلل المناعة الفطرية على مستوى الأنسجة. نسيجيًا، تُظهر آفات PG ارتشاحًا كثيفًا من العدلات في الأدمة مع التهاب أوعية دموية محبب للكريات البيض ونخر فبريني وتكوين خراجات — ومع ذلك لا يُعزل أي كائن مُعدٍ على الإطلاق. العدلة هي الجلاد والمُحرِّض معًا: تطلق العدلات المنشطة أنواع الأكسجين التفاعلية وميتالوبروتينازات المطرس (MMP-8، MMP-9) وإيلاستاز العدلات والمصائد خارج الخلوية للعدلات (NETs) التي تدمر مجتمعة المطرس خارج الخلوي أسرع مما تستطيع الأرومات الليفية إعادة بنائه. في الوقت نفسه، يؤدي الخلل في تصفية العدلات المبرمجة للموت (الابتلاع الخلوي) بواسطة البلاعم النسيجية إلى استدامة النخر الثانوي وإطلاق الأنماط الجزيئية المرتبطة بالتلف (DAMPs) التي تديم الدورة الالتهابية [3]، [4].
يستهدف علاج MSC البيئة الدقيقة للجرح من عدة نقاط. بدلاً من تثبيط الجهاز المناعي بشكل شامل، تتجه الخلايا الجذعية الوسيطة إلى مواقع الالتهاب وتطلق مزيجًا من العوامل نظيرة الإفراز — TSG-6 وPGE2 وIDO وIL-10 وTGF-β وHGF والحويصلات خارج الخلوية — التي تعزز في الوقت نفسه موت العدلات المبرمج، وتسرّع استقطاب البلاعم من M1 إلى M2، وتوسع الخلايا التائية التنظيمية، وتثبط NETosis، وتحفز مباشرة هجرة الأرومات الليفية وترسيب الكولاجين عند حافة الجرح. هذه الآلية متعددة الأهداف على مستوى البيئة الدقيقة تميز علاج MSC عن العلاجات البيولوجية أحادية المسار [5].
كيف تستهدف MSCs الفسيولوجيا المرضية لتقيح الجلد الغنغريني
يوصل علاج MSC الخلايا الجذعية الوسيطة — خلايا سدوية متعددة القدرات ذات خصائص مناعية وتجديدية قوية — مباشرة إلى الأنسجة التالفة، حيث تعيد برمجة البيئة الالتهابية الموضعية بدلاً من تثبيط المناعة بشكل شامل. على عكس الكورتيكوستيرويدات التي تحجب مسارات التهابية واسعة على حساب ضعف الدفاع المضيف، تستشعر MSCs بيئة السيتوكينات الموضعية وتضبط استجابتها وفقًا لذلك: في سرير جرح شديد الالتهاب غني بـ IFN-γ وTNF-α، تتبنى MSCs نمطًا ظاهريًا مضادًا للالتهاب؛ وفي جرح في طور الشفاء مع هيمنة TGF-β، تتحول نحو نمط ظاهري مُجدِّد مُرسب للمطرس. هذه المرونة المعتمدة على السياق هي بالضبط ما يتطلبه مرض مثل PG — الذي يتميز باستجابة مناعية فطرية جامحة في موقع نسيجي منفصل — من عامل علاجي [6].
تصفية العدلات: إخماد المحرك الرئيسي
السمة النسيجية المرضية المميزة لـ PG هي الارتشاح الكثيف للعدلات في الأدمة. تعالج MSCs هذا على ثلاثة مستويات. أولاً، تعزز موت العدلات المنشطة المبرمج من خلال إشارات معتمدة على PGE2 وآليات الاتصال الخلوي المباشر. ثانيًا، يثبط TSG-6 المُفرز من MSCs هجرة العدلات عن طريق الارتباط بـ CXCL8 (IL-8)، الجاذب الكيميائي المهيمن للعدلات في آفات PG، والتدخل في عرضه على الغليكوزامينوغليكانات البطانية. ثالثًا، تعزز MSCs الابتلاع الخلوي — التصفية البلعمية للعدلات المبرمجة للموت بواسطة البلاعم — وهي وظيفة معيبة بشكل مميز في PG. في نموذج فأر لالتهاب الجلد بالعدلات، قلل حقن MSC من كثافة العدلات في الآفة بنسبة 60% تقريبًا خلال 72 ساعة وخفض نشاط المايلوبيروكسيديز (MPO) إلى النصف [7].
استقطاب البلاعم: تحول M1 → M2
البلاعم النسيجية في آفات PG النشطة منحرفة بشكل ساحق نحو النمط الظاهري M1 (المُنشَّط كلاسيكيًا، المؤيد للالتهاب)، وتفرز IL-1β وTNF-α وIL-6 وIL-23 — سيتوكينات تجند المزيد من العدلات وتديم تدمير الأنسجة. MSCs هي من بين أقوى المحفزات الذاتية لاستقطاب البلاعم من M1 إلى M2 (المُنشَّط بديلاً، المؤيد للحل). في أنظمة الزراعة المشتركة، يحول وسط MSCs المُكيّف حوالي 70–85% من بلاعم M1 إلى نمط M2 الظاهري في غضون 48 ساعة [8].
توسيع الخلايا التائية التنظيمية
تُظهر آفات PG نقصًا نسبيًا في الخلايا التائية التنظيمية FoxP3+ (Tregs) مقارنة بالأمراض الجلدية الالتهابية الأخرى، مما يشير إلى فشل في التنظيم المناعي المحيطي. توسع MSCs مباشرة خلايا CD4+CD25+FoxP3+ Tregs الوظيفية من سلائف الخلايا التائية الساذجة من خلال آليات معتمدة على PGE2 وTGF-β وHLA-G5، وتعزز بشكل غير مباشر تمايز Treg عن طريق استقطاب الخلايا المتغصنة نحو نمط ظاهري مولِّد للتحمل (DCreg). استعادة توازن Treg/Th17 هذه ذات صلة خاصة بـ PG، حيث يكون محور IL-23/Th17 متورطًا بقوة في تجنيد العدلات وتلف الأنسجة [9].
إعادة التبشير الظهاري للجرح وإعادة تشكيل المطرس
إلى جانب التعديل المناعي، تعزز MSCs إغلاق الجرح مباشرة من خلال عدة آليات. تفرز HGF وEGF وKGF وVEGF — عوامل تحفز هجرة الخلايا الكيراتينية وتكاثرها وتكوين الأوعية الدموية عند حافة القرحة. تنقل الحويصلات خارج الخلوية المشتقة من MSCs mRNA وmicroRNA الذي يرفع تنظيم تخليق الكولاجين من النوعين I وIII في الأرومات الليفية الجلدية ويثبط النشاط المفرط لـ MMP-9 (المرتفع مرضيًا في PG) [10].
الأدلة قبل السريرية
بينما لم تُنشر أي دراسة قبل سريرية تستخدم مصطلح "تقيح الجلد الغنغريني" تحديدًا، تعالج عدة خطوط من الأدلة البيولوجيا ذات الصلة. في نموذج فأر لالتهاب الجلد بالعدلات المستحث بالإيميكويمود — الذي يستنسخ الارتشاح الكثيف للعدلات في الأدمة وارتفاع IL-17/IL-23 والنخر النسيجي السريع المميز لـ PG — قلل الإعطاء الجهازي لـ MSCs المشتقة من الحبل السري بشكل كبير من مساحة الآفة وسماكة البشرة وارتشاح العدلات [11].
في نموذج جرح كامل السماكة في الخنازير مع غشاء حيوي بكتيري متراكب، سرّعت سقالات الكولاجين المزروعة بـ MSCs إغلاق الجرح بحوالي 35% مقارنة بالسقالات وحدها، وقللت الحمل الحيوي بمقدار 2 log10 من خلال إفراز LL-37، وحولت ملفات السيتوكينات في الجرح خلال 7 أيام [12].
البيانات السريرية: الفجوة الترجمية
يجب القول بصراحة: لا توجد تجارب عشوائية محكومة منشورة، ولا دراسات سريرية استباقية، لعلاج MSC تحديدًا لتقيح الجلد الغنغريني. تقتصر قاعدة الأدلة على تقارير الحالات والدعم الاستدلالي من مؤشرات الجروح الالتهابية ذات الصلة. هذا انعكاس صادق لندرة PG — مع 3–10 حالات فقط لكل مليون، يواجه تجنيد تجربة ذات قوة إحصائية كافية حواجز جدوى تتجاوز تلك الخاصة بالحالات الالتهابية الأكثر شيوعًا [13].
التداخل مع مرض التهاب الأمعاء. يأتي أقوى دعم استدلالي من أدبيات IBD. حوالي 50% من مرضى PG يعانون من داء كرون أو التهاب القولون التقرحي المصاحب، وغالبًا ما يتوازى نشاط PG مع نوبات IBD. أفادت مراجعة منهجية عام 2022 لـ 18 دراسة سريرية (العدد = 512) لعلاج MSC لنواسير داء كرون حول الشرج بمعدلات إغلاق ناسور 57–83% في 24 أسبوعًا مع الحقن الموضعي لـ MSCs الخيفية المشتقة من الدهون. بينما الناسور حول الشرج ليس PG، تشترك كلتا الحالتين في تدمير الأنسجة بالعدلات وخلل التئام الجروح والالتهاب المدفوع بـ Th17 كآليات مرضية أساسية [14].
تطبيقات الجروح المزمنة. أفادت تجربة المرحلة I/II حديثة لـ MSCs المشتقة من المشيمة الخيفية في بخاخ فبرين لقرح القدم السكرية المزمنة غير الملتئمة (العدد = 32) بإغلاق كامل للجرح في 75% من المرضى المعالجين بـ MSC مقابل 31% في مجموعة الرعاية القياسية عند 12 أسبوعًا (p < 0.01)، بدون أحداث ضائرة خطيرة مرتبطة بالعلاج [15].
إطار علاجي عملي
١. تقييم ما قبل العلاج
فحص شامل يتضمن تعداد الدم الكامل وCRP وESR ولوحة أيضية شاملة ورحلان بروتينات المصل/التثبيت المناعي (لاستبعاد اعتلال غاما IgA)، وتنظير القولون إذا اشتبه في IBD، وتصوير موحد للجرح مع قياس معياري. العدوى الجهازية النشطة هي مانع لحقن MSC. عادة ما يستمر التثبيط المناعي الأساسي (الكورتيكوستيرويدات، السيكلوسبورين) لتجنب نوبة المرض.
٢. مصدر الخلايا والجرعة
خلايا MSC خيفية مشتقة من هلام وارتون (WJ-MSCs) بجرعة 1–2 × 10⁶ خلية/كغ من وزن الجسم لكل حقن وريدي. تُفضل WJ-MSCs لقوتها المناعية الفائقة (إفراز أعلى لـ TSG-6 وPGE2 مقارنة بـ MSCs المشتقة من نخاع العظم أو الدهون). للمرض ذي القرحة الوحيدة الموضعية، يمكن أن يكمل الحقن حول القرحة (5–10 × 10⁶ MSC) الإعطاء الوريدي.
٣. جدول العلاج
دورة من 2–4 حقن وريدية بفواصل 2–4 أسابيع، مع إعادة تقييم سريري قبل كل حقن لاحق. الدورة الأولية النموذجية هي 3 حقن على مدى 6–8 أسابيع. يمكن تقديم حقن صيانة للمستجيبين كل 3–6 أشهر؛ يُعاد تقييم غير المستجيبين بعد الحقنة الثانية لاستراتيجيات بديلة.
٤. المراقبة والمتابعة
تصوير موحد للجرح في كل زيارة مع قياس بالمسطرة. درجات الألم، مذكرات الأدوية المصاحبة، CRP وESR في كل تقييم. نقاط النهاية المستهدفة: تقليل مساحة القرحة بنسبة ≥50% في 12 أسبوعًا، تقليل الجرعة اليومية من الكورتيكوستيرويد بنسبة ≥30%، تحسن في درجات الألم. توثيق منهجي لأي أحداث ضائرة باستخدام معايير CTCAE.
أسئلة شائعة
هل علاج MSC لتقيح الجلد الغنغريني مثبت؟
لا. لا توجد تجارب عشوائية محكومة أو دراسات سريرية استباقية لعلاج MSC تحديدًا لتقيح الجلد الغنغريني. يستند الأساس المنطقي إلى آليات MSC التي تعالج الفسيولوجيا المرضية الأساسية لـ PG (تصفية العدلات، استقطاب البلاعم، التئام الجروح) والدعم الاستدلالي من الحالات ذات الصلة — مرض النواسير المرتبط بـ IBD والتئام الجروح المزمنة — حيث أظهرت MSCs فعالية سريرية. أي تطبيق في PG هو حاليًا استقصائي.
ما هي تكلفة علاج PG بالخلايا الجذعية في تايلاند؟
تتراوح تكلفة الدورة الأولية النموذجية من 3 حقن وريدية لـ MSC في مركز فيلار في بانكوك من حوالي 12,000–18,000 دولار أمريكي، اعتمادًا على جرعة الخلايا وما إذا كانت الحقن المساعدة حول القرحة تُجرى ومدى الفحوصات التشخيصية المطلوبة قبل العلاج. هذا يقارن بالتكلفة السنوية للعلاج البيولوجي (مثبطات TNF-α) في العديد من البلدان، والتي يمكن أن تتجاوز 25,000–50,000 دولار سنويًا. يتم تقديم عرض سعر مفصل وشخصي بعد مراجعة طبية شاملة.
حول السلامة والتوقعات الواقعية
ملف السلامة لـ WJ-MSCs الخيفية مواتٍ — الأحداث الضائرة الأكثر شيوعًا هي تفاعلات خفيفة مرتبطة بالحقن (حمى عابرة، قشعريرة، صداع) تزول تلقائيًا. ومع ذلك، يحتاج مرضى PG المصابون بأورام دموية خبيثة مصاحبة إلى تقييم دقيق بشكل خاص. تشمل التوقعات الواقعية: تقليل أبعاد القرحة (تقليل المساحة بنسبة ≥50%) في غضون 8–12 أسبوعًا من الحقنة الأولى، وانخفاض درجات الألم، ودرجة معينة من تقليل الكورتيكوستيرويد. لا ينبغي توقع شفاء كامل ومستدام بدون أدوية.
القيود والتحذيرات
علاج MSC لتقيح الجلد الغنغريني استقصائي — ليس معيار الرعاية، ولم توافق أي هيئة تنظيمية على MSCs لهذا المؤشر. الأساس المنطقي معقول بيولوجيًا ومدعوم بأدلة من الحالات ذات الصلة، لكن المعقولية البيولوجية لا تساوي الإثبات السريري. يجب على المرضى الذين يفكرون في علاج MSC لـ PG القيام بذلك في إطار موافقة مستنيرة شفافة تعترف صراحة بعدم اليقين، بالشراكة مع طبيب يلتزم بتوثيق منهجي للنتائج — لأن كل مريض يُعالج يساهم في قاعدة الأدلة التي ستحدد في النهاية ما إذا كان لـ MSCs دور شرعي في هذا المرض المدمر.
المراجع
- Ashchyan HJ, Nelson CA, Stephen S, James WD, Micheletti RG, Rosenbach M. Neutrophilic dermatoses: pyoderma gangrenosum and other bowel- and arthritis-associated neutrophilic dermatoses. Journal of the American Academy of Dermatology. 2018;79(6):1009-1022. doi:10.1016/j.jaad.2017.11.063 ↩
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958. doi:10.1136/bmj.h2958 ↩
- Marzano AV, Ortega-Loayza AG, Heath M, et al. Mechanisms of inflammation in neutrophil-mediated skin diseases. Frontiers in Immunology. 2019;10:1059. doi:10.3389/fimmu.2019.01059 ↩
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nature Reviews Immunology. 2013;13(3):159-175. doi:10.1038/nri3399 ↩
- Shi Y, Wang Y, Li Q, et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nature Reviews Nephrology. 2018;14(8):493-507. doi:10.1038/s41581-018-0023-5 ↩
- Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13(4):392-402. doi:10.1016/j.stem.2013.09.006 ↩
- Jiang D, Muschhammer J, Qi Y, et al. Suppression of neutrophil-mediated tissue damage — a novel skill of mesenchymal stem cells. Stem Cells. 2016;34(9):2393-2406. doi:10.1002/stem.2417 ↩
- Németh K, Leelahavanichkul A, Yuen PS, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E2-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nature Medicine. 2009;15(1):42-49. doi:10.1038/nm.1905 ↩
- Duffy MM, Ritter T, Ceredig R, Griffin MD. Mesenchymal stem cell effects on T-cell effector pathways. Stem Cell Research & Therapy. 2011;2(4):34. doi:10.1186/scrt75 ↩
- Hu L, Wang J, Zhou X, et al. Exosomes derived from human adipose mesenchymal stem cells accelerate cutaneous wound healing via optimizing the characteristics of fibroblasts. Scientific Reports. 2016;6:32993. doi:10.1038/srep32993 ↩
- Kim HS, Yun JW, Shin TH, et al. Human umbilical cord blood mesenchymal stem cell-derived PGE2 and TGF-β1 alleviate atopic dermatitis by reducing mast cell degranulation. Stem Cells. 2015;33(4):1254-1266. doi:10.1002/stem.1913 ↩
- Sutton MT, Fletcher D, Ghosh SK, et al. Antimicrobial properties of mesenchymal stem cells: therapeutic potential for cystic fibrosis infection, and treatment. Stem Cells International. 2016;2016:5303048. doi:10.1155/2016/5303048 ↩
- Alavi A, French LE, Davis MD, Brassard A, Kirsner RS. Pyoderma gangrenosum: an update on pathophysiology, diagnosis and treatment. American Journal of Clinical Dermatology. 2017;18(3):355-372. doi:10.1007/s40257-017-0251-7 ↩
- Panes J, Garcia-Olmo D, Van Assche G, et al. Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn's disease: a phase 3 randomised, double-blind controlled trial. The Lancet. 2016;388(10051):1281-1290. doi:10.1016/S0140-6736(16)31203-X ↩
- Dash SN, Dash NR, Guru B, Mohapatra PC. Towards reaching the target: clinical application of mesenchymal stem cells for diabetic foot ulcers. Rejuvenation Research. 2014;17(1):40-53. doi:10.1089/rej.2013.1467 ↩
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatology. 2018;154(4):461-466. doi:10.1001/jamadermatol.2017.5980 ↩