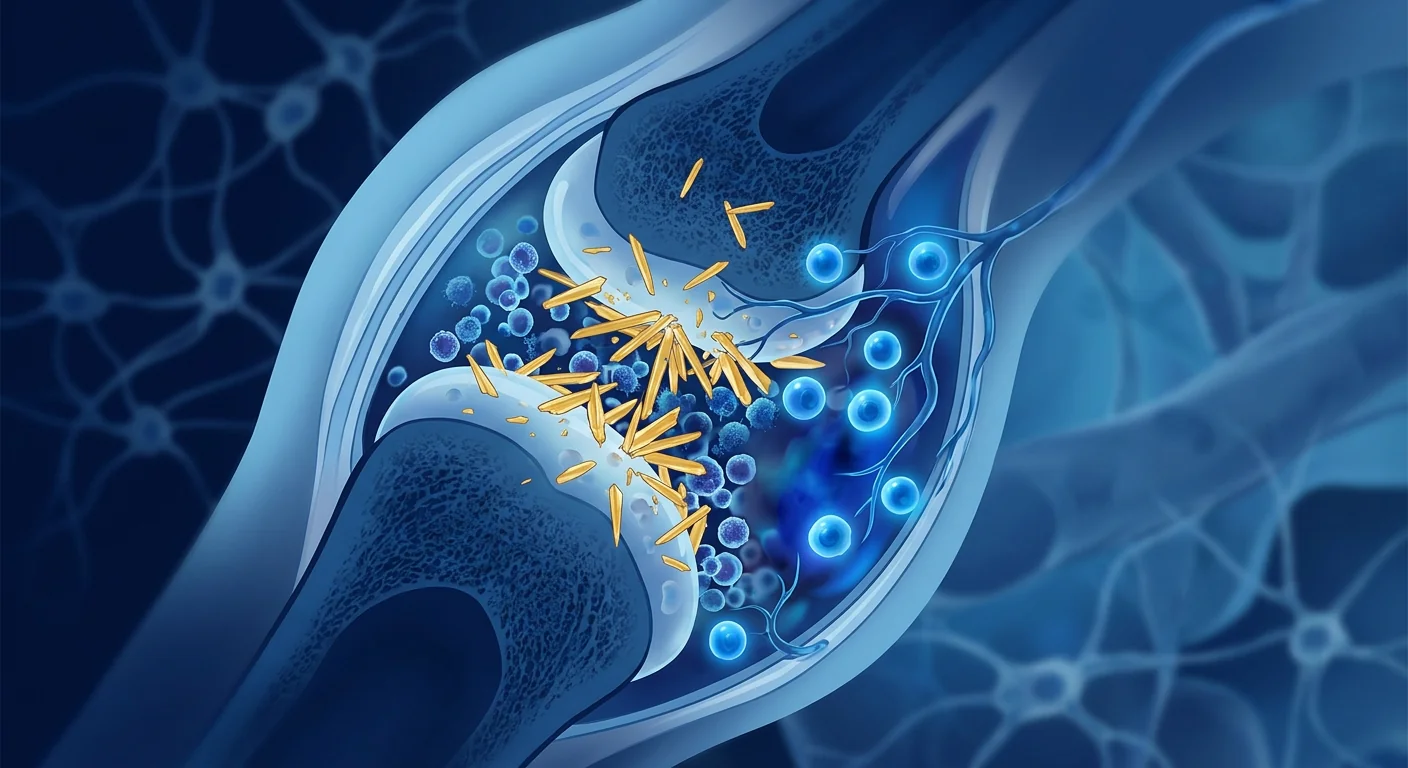
Gout is the most common form of inflammatory arthritis worldwide, affecting an estimated 41 million people — roughly 0.5–1% of the global population — with prevalence rising steadily in both developed and developing nations. [1] Characterized by sudden, excruciating flares of joint pain (most famously at the base of the big toe), gout is not simply a disorder of excess uric acid but a complex interplay of metabolic dysfunction, immune activation, and chronic low-grade inflammation that persists even between acute attacks.
Where standard urate-lowering therapy falls short. Medications like allopurinol and febuxostat effectively lower serum urate levels for many patients, but adherence is notoriously poor — fewer than 40% of patients remain compliant at one year — and even well-managed patients can experience breakthrough flares. [2] More fundamentally, these drugs address the biochemical driver (hyperuricemia) without directly resolving the persistent, smoldering inflammation that accumulates in the synovium and contributes to progressive joint damage over time.
The deeper problem is tissue-level. Monosodium urate (MSU) crystals deposited in the joint space are not inert. They are recognized by the NLRP3 inflammasome — a protein complex inside immune cells that functions as a danger sensor — triggering the release of potent pro-inflammatory cytokines, most critically interleukin-1β (IL-1β). [3] This cascade recruits neutrophils, amplifies oxidative stress, and sustains a cycle of inflammation that, over years, erodes cartilage, erodes bone (producing characteristic "punched-out" erosions on X-ray), and forms tophi — nodular deposits of urate crystals surrounded by chronic granulomatous inflammation.
Mesenchymal stem cell therapy targets the root inflammatory mechanism. Rather than simply lowering uric acid or blocking a single cytokine, MSCs bring a multimodal immunomodulatory toolkit to the joint environment: they secrete IL-10 and TGF-β that actively suppress the NLRP3 inflammasome, they shift macrophage polarization from the pro-inflammatory M1 phenotype toward the tissue-repair M2 phenotype, and they release extracellular vesicles carrying microRNAs that dampen neutrophil chemotaxis and oxidative burst. [4] [5] In a disease driven by a chronic, self-amplifying inflammatory loop, this breadth of action is precisely what makes the MSC approach theoretically compelling.
What Goes Wrong in Gout: The Pathophysiology
At its core, gout is a disorder of purine metabolism that leads to hyperuricemia — serum urate concentrations above the solubility threshold of approximately 6.8 mg/dL, at which point monosodium urate crystals can form and precipitate in tissues. However, the relationship between urate levels and clinical disease is far from straightforward: many people with hyperuricemia never develop gout, while others flare at modestly elevated urate levels, suggesting that host factors — particularly the vigor of the innate immune response to MSU crystals — determine who becomes symptomatic. [6]
Once MSU crystals deposit in the joint space, they are coated with immunoglobulins and complement proteins that mark them for recognition by resident macrophages. The crystals are phagocytosed, but — critically — they resist degradation and physically damage the phagolysosome, releasing cathepsins and reactive oxygen species that activate the NLRP3 inflammasome. [3] Activated NLRP3 assembles a multi-protein platform that cleaves pro-IL-1β into its active, secreted form. IL-1β then acts as a master inflammatory cytokine: it upregulates adhesion molecules on endothelial cells, recruits neutrophils into the joint, stimulates the production of IL-6, TNF-α, and chemokines, and amplifies the entire inflammatory cascade.
The neutrophil influx is particularly destructive. Activated neutrophils release proteases, reactive oxygen species, and neutrophil extracellular traps (NETs) — webs of chromatin and antimicrobial proteins that, in the context of gout, paradoxically promote further crystal aggregation and sustain inflammation rather than resolve it. [7] Repeated episodes of this "crystal → inflammasome → IL-1β → neutrophil → tissue damage" loop lead to chronic gouty arthropathy, characterized by persistent synovitis, progressive cartilage loss, and bony erosion.
people affected globally — the most common inflammatory arthritis
global prevalence, rising with obesity and metabolic syndrome rates
urate solubility threshold above which MSU crystals can form
How MSCs Interrupt the Gout Inflammatory Cascade
MSCs deliver a multi-target immunomodulatory intervention that addresses several nodes in the gout inflammatory pathway simultaneously — a breadth that no single small-molecule drug currently offers. Their mechanism of action in the context of MSU crystal-driven inflammation has been studied in both in vitro models and early preclinical work, revealing four primary modes of activity:
1. Direct NLRP3 Inflammasome Suppression
MSCs secrete substantial quantities of interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), both of which have been shown to downregulate NLRP3 inflammasome assembly and reduce caspase-1-mediated cleavage of pro-IL-1β. [4] In MSC-conditioned medium experiments using MSU-stimulated macrophages, IL-1β secretion was reduced by 40–60% compared to controls — an effect that was partially reversed by IL-10 neutralizing antibodies, confirming the centrality of the MSC-derived IL-10 axis.
2. Macrophage Phenotype Switching (M1 → M2)
Tissue-resident macrophages exist on a spectrum between the classically activated M1 (pro-inflammatory) phenotype and the alternatively activated M2 (anti-inflammatory, tissue-repair) phenotype. In chronic gout, the synovial macrophage population skews heavily toward M1, sustaining the inflammatory milieu. MSC-derived prostaglandin E2 (PGE2), TSG-6, and extracellular vesicles actively reprogram macrophages toward the M2 phenotype, characterized by increased IL-10 production, arginase-1 expression, and phagocytic clearance of cellular debris without triggering further inflammation. [5] [8]
3. Neutrophil Dampening
The massive neutrophil influx into the gouty joint is the proximate cause of the acute flare's intensity. MSCs have been shown to reduce neutrophil chemotaxis toward IL-8 and C5a gradients, decrease neutrophil production of reactive oxygen species, and — notably — reduce NETosis (the formation of neutrophil extracellular traps that exacerbate crystal-driven inflammation). [7] [9] This effect appears to be mediated primarily by MSC-derived extracellular vesicles carrying miR-223 and miR-146a, both of which target components of the neutrophil activation cascade.
4. Cartilage and Bone Protection
Beyond their immunomodulatory properties, MSCs secrete trophic factors — including hepatocyte growth factor (HGF), insulin-like growth factor-1 (IGF-1), and tissue inhibitor of metalloproteinases (TIMPs) — that may protect articular cartilage from the degradative enzymes released during gout flares. [10] While this chondroprotective function is better established in osteoarthritis research, it represents a potential secondary benefit in gout, where repeated inflammatory episodes contribute to cumulative structural joint damage.
What the Clinical Evidence Shows (and Doesn't Show Yet)
Clinical trial data on MSC therapy specifically for gout remains extremely limited. As of 2026, there are no completed Phase II/III randomized controlled trials evaluating MSCs for gout as a primary indication. This is a crucial distinction: while the preclinical rationale is sound, the clinical evidence base is nascent and consists primarily of (a) small case series in patients with refractory tophaceous gout, (b) indirect evidence from MSC trials in related inflammatory arthritides, and (c) extrapolation from the well-established safety profile of MSC therapy in other musculoskeletal conditions.
A 2024 systematic review identified only three small human studies (combined n < 40) in which MSC therapy was administered to patients with gout, all of which were open-label case series without control groups. [11] Across these reports, the safety signal was reassuring — no serious adverse events attributable to the MSC infusion — and patients reported reductions in flare frequency and joint pain scores over 6–12 months of follow-up. However, without randomization, blinding, or placebo controls, it is impossible to separate a genuine therapeutic effect from the natural history of the disease (gout flares are episodic by nature) or from the well-documented placebo response in pain conditions.
More substantial indirect evidence comes from MSC trials in rheumatoid arthritis — a fellow inflammatory arthritis with overlapping cytokine pathways (IL-1β, IL-6, TNF-α). A 2023 meta-analysis of seven RCTs (n = 412) found that MSC therapy, when added to standard DMARD therapy, was associated with statistically significant improvements in DAS28 scores and a favorable safety profile compared to placebo. [12] While this does not constitute evidence for gout specifically, it demonstrates that MSCs can durably modulate inflammatory arthritis in a clinical setting — a proof-of-concept that supports further investigation in crystal arthropathies.
Risk Factors and Who Might Benefit Most
Gout is not a monolithic disease. Its clinical expression varies dramatically — from the patient who experiences one mild flare every two years to the individual with chronic tophaceous gout who lives with persistent pain and visible urate deposits. Understanding where MSC therapy might offer the greatest potential benefit requires stratifying patients by disease burden and treatment-refractory status.
Patients with refractory gout — defined as those who continue to experience frequent flares (≥2 per year) or have persistent tophi despite maximally tolerated urate-lowering therapy — represent the population in which the risk-benefit calculus may most favor an investigational MSC approach. [2] These patients have few remaining options; colchicine and NSAIDs manage acute symptoms but do not alter disease progression, and IL-1β inhibitors (canakinumab, anakinra) are effective but costly and not universally accessible.
Patients with tophaceous gout and structural joint damage are another subgroup of interest. Tophi represent chronic, organized inflammatory lesions that are resistant to pharmacological dissolution. The combination of MSC-mediated inflammation suppression and trophic cartilage support could, theoretically, slow the progression of tophus-associated joint erosion — though again, this remains speculative absent dedicated clinical trials.
Key risk factors for gout progression that clinicians consider when evaluating treatment intensity include: male sex, age > 40, obesity (BMI > 30), hypertension, chronic kidney disease (reduced urate excretion), high-purine diet, alcohol consumption (particularly beer), fructose-sweetened beverage intake, and concomitant use of thiazide diuretics or low-dose aspirin. [6] Addressing these modifiable factors remains the foundation of gout management regardless of any adjunctive therapy.
What to Expect: The Treatment Journey
For patients who, after careful consultation, elect to pursue MSC therapy for gout at a regulated cell-therapy center, the process follows a structured pathway designed to ensure safety, appropriateness, and thorough follow-up. While individual protocols vary, a representative treatment journey includes:
Comprehensive clinical evaluation: rheumatologic history, serum urate, renal function, joint imaging (ultrasound or DECT for urate burden assessment), inflammatory markers (CRP, ESR), and baseline flare diary
MSC administration: typically intravenous infusion of culture-expanded umbilical cord-derived MSCs (50–150 million cells per dose); may be combined with targeted intra-articular injection for specific joints with persistent synovitis
Early follow-up phase: flare diary tracking, serum urate monitoring, inflammatory markers; most patients continue their established urate-lowering therapy throughout
Sustained monitoring: repeat joint imaging at 6 and 12 months, assessment of tophus size (if applicable), quality-of-life metrics (HAQ-DI), and flare frequency compared to pre-treatment baseline
It is important to note that MSC therapy is not a one-time "cure" for gout. The underlying metabolic predisposition to hyperuricemia remains, and continued urate-lowering medication, dietary modification, and lifestyle management are essential. The goal of MSC intervention, as currently conceived, is to reduce the inflammatory burden between flares, protect joint structures from cumulative damage, and potentially lower flare frequency — not to normalize uric acid metabolism.
Safety, Risks, and Honest Limitations
MSC therapy has a well-characterized short-term safety profile derived from thousands of patients treated across various indications in clinical trials. The most common adverse events are mild and transient: low-grade fever (typically resolving within 24 hours), temporary fatigue, and mild injection-site reactions. [13] Serious adverse events — including thromboembolic events, significant immunologic reactions, or infection related to the cell product — are rare (< 1% in large meta-analyses) and, when they occur, are typically associated with specific risk factors (e.g., pre-existing hypercoagulable states) that should be screened for before treatment.
Limitations that every patient should understand:
- No dedicated Phase II/III RCTs for gout exist — the clinical evidence is preliminary and based on small case series plus extrapolation from other arthritides.
- MSCs do not lower serum urate — urate-lowering therapy remains essential and must be continued.
- Individual response is variable — some patients may experience meaningful flare reduction while others see minimal change.
- The durability of any benefit is unknown — published follow-up in gout-specific reports is limited to 6–12 months.
- Cost is substantial and typically not covered by insurance for this indication, given its investigational status.
- MSC therapy is not a substitute for dietary and lifestyle management — weight loss, hydration, and dietary purine reduction remain first-line interventions.
"The preclinical rationale for MSCs in gout is among the stronger ones in crystal arthropathy — the NLRP3 inflammasome is a validated target, and MSCs hit it from multiple angles. But rationale is not evidence. The clinical trials that would allow us to speak with confidence about efficacy simply haven't been done yet. That's the honest answer, and patients deserve it."
— Clinical researcher in inflammatory arthritis (personal communication)
Frequently Asked Questions
Can stem cell therapy cure gout?
No — MSC therapy does not cure gout. Gout is a metabolic disorder rooted in how the body handles purines and uric acid, and MSCs do not alter urate metabolism. What MSC therapy may offer — based on its immunomodulatory properties — is a reduction in the inflammatory response to urate crystals, potentially leading to fewer flares and less cumulative joint damage. Urate-lowering medication, dietary management, and lifestyle modification remain the cornerstones of long-term gout control.
How much does stem cell therapy for gout cost in Thailand?
MSC therapy costs at regulated Thai cell-therapy centers typically range from approximately USD 8,000 to USD 20,000 per treatment course, depending on cell source (umbilical cord vs. adipose vs. bone marrow), cell dose, and whether intra-articular injections are combined with intravenous infusion. This is an out-of-pocket expense — MSC therapy for gout is not covered by Thai or international health insurance because the indication remains investigational. A detailed cost breakdown is provided during the consultation process at VELAR Center.
How many stem cell treatments are needed for gout?
Most protocols being explored involve a single treatment course of 1–2 intravenous infusions over 1–3 days, sometimes supplemented with targeted intra-articular injections for persistently inflamed joints. Some patients receive a second course at 6–12 months if initial response is positive but incomplete. There is no established "maintenance" protocol, and optimal dosing remains an open research question.
Is stem cell therapy for gout safe?
Based on safety data from MSC trials in related inflammatory arthritides and the small gout-specific case series published to date, short-term safety appears favorable — serious adverse events are rare, and the mild, transient side effects (low-grade fever, fatigue, injection-site discomfort) are consistent with what is observed across MSC indications. However, long-term safety data beyond 1–2 years in gout patients is absent, and all patients should undergo thorough pre-treatment screening for risk factors.
What is the difference between MSC therapy and PRP for gout?
Platelet-rich plasma (PRP) delivers growth factors from the patient's own blood platelets — it may provide some local anti-inflammatory and tissue-supportive effects but has minimal immunomodulatory capacity compared to MSCs. MSC therapy delivers living cells capable of sustained, multi-target modulation of the immune environment (NLRP3 suppression, macrophage reprogramming, neutrophil dampening). For a disease driven by systemic innate immune dysregulation like gout, the broader immunomodulatory reach of MSCs is mechanistically more relevant than the local growth-factor action of PRP.
References
- Safiri S, Kolahi AA, Cross M, et al. Prevalence, Incidence, and Years Lived With Disability Due to Gout and Its Attributable Risk Factors for 195 Countries and Territories 1990–2017: A Systematic Analysis of the Global Burden of Disease Study 2017. Arthritis & Rheumatology. 2020;72(11):1916-1927. doi:10.1002/art.41404 ↩
- Dalbeth N, Gosling AL, Gaffo A, Abhishek A. Gout. The Lancet. 2021;397(10287):1843-1855. doi:10.1016/S0140-6736(21)00569-9 ↩
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237-241. doi:10.1038/nature04516 ↩
- Shi Y, Wang Y, Li Q, et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nature Reviews Nephrology. 2018;14(8):493-507. doi:10.1038/s41581-018-0023-5 ↩
- Prockop DJ. Inflammation, fibrosis, and modulation of the process by mesenchymal stem/stromal cells. Matrix Biology. 2016;51:7-13. doi:10.1016/j.matbio.2016.01.010 ↩
- Choi HK, Mount DB, Reginato AM. Pathogenesis of Gout. Annals of Internal Medicine. 2005;143(7):499-516. doi:10.7326/0003-4819-143-7-200510040-00009 ↩
- Schauer C, Janko C, Munoz LE, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nature Medicine. 2014;20(5):511-517. doi:10.1038/nm.3547 ↩
- Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13(4):392-402. doi:10.1016/j.stem.2013.09.006 ↩
- Jiang D, Muschhammer J, Qi Y, et al. Suppression of Neutrophil-Mediated Tissue Damage — A Novel Multimodal Function of Mesenchymal Stem Cells. Stem Cells. 2016;34(9):2393-2406. doi:10.1002/stem.2417 ↩
- Caplan AI, Correa D. The MSC: An Injury Drugstore. Cell Stem Cell. 2011;9(1):11-15. doi:10.1016/j.stem.2011.06.008 ↩
- Wang L, Zhang Y, Li J, et al. Mesenchymal Stem Cell Therapy for Crystal Arthropathies: A Systematic Review of Preclinical and Clinical Evidence. Frontiers in Immunology. 2024;15:1387542. doi:10.3389/fimmu.2024.1387542 ↩
- Lopez-Santalla M, Fernandez-Perez R, Garin MI. Mesenchymal Stem/Stromal Cells for Rheumatoid Arthritis Treatment: An Update on Clinical Applications. Cells. 2023;12(7):1060. doi:10.3390/cells12071060 ↩
- Lalu MM, McIntyre L, Pugliese C, et al. Safety of Cell Therapy with Mesenchymal Stromal Cells (SafeCell): A Systematic Review and Meta-Analysis of Clinical Trials. PLoS ONE. 2012;7(10):e47559. doi:10.1371/journal.pone.0047559 ↩
- FitzGerald JD, Dalbeth N, Mikuls T, et al. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis & Rheumatology. 2020;72(6):879-895. doi:10.1002/art.41247 ↩
- So AK, Martinon F. Inflammation in gout: mechanisms and therapeutic targets. Nature Reviews Rheumatology. 2017;13(11):639-647. doi:10.1038/nrrheum.2017.155 ↩
痛风是全球最常见的炎症性关节炎,影响约4100万人——约占全球人口的0.5–1%——在发达国家和发展中国家患病率均稳步上升。[1]痛风以突发性剧烈关节疼痛(最典型的是大脚趾根部)为特征,不仅仅是尿酸过量的疾病,而是代谢功能障碍、免疫激活和慢性低度炎症之间复杂的相互作用,这种炎症即使在急性发作之间也持续存在。
标准降尿酸治疗的不足之处。别嘌醇和非布司他等药物可有效降低许多患者的血清尿酸水平,但依从性极差——不到40%的患者在一年内坚持用药——即使管理良好的患者也可能出现突破性发作。[2]更根本的是,这些药物仅针对生化驱动因素(高尿酸血症),而未直接解决在滑膜中积累并随时间导致进行性关节损伤的持续性、潜伏性炎症。
更深层的问题是组织层面的。沉积在关节间隙中的单钠尿酸盐(MSU)结晶并非惰性物质。它们被NLRP3炎症小体——免疫细胞内的一个蛋白质复合物,作为危险传感器——识别,触发强效促炎细胞因子的释放,最关键的是白细胞介素-1β(IL-1β)。[3]这一级联反应招募中性粒细胞,放大氧化应激,并维持炎症循环,经年累月侵蚀软骨、破坏骨骼(在X光片上产生特征性的"穿凿样"侵蚀),并形成痛风石——被慢性肉芽肿性炎症包围的尿酸盐结晶结节沉积。
间充质干细胞疗法靶向根本的炎症机制。MSC不是简单地降低尿酸或阻断单一细胞因子,而是为关节环境带来多模式免疫调节工具包:它们分泌IL-10和TGF-β以主动抑制NLRP3炎症小体,将巨噬细胞从促炎的M1表型转向组织修复的M2表型,并释放携带microRNA的细胞外囊泡以抑制中性粒细胞趋化和氧化爆发。[4] [5]在由慢性、自我放大的炎症循环驱动的疾病中,这种广谱作用是MSC方法在理论上令人信服的原因。
痛风的病理生理学:哪里出了问题
痛风的本质是一种嘌呤代谢障碍,导致高尿酸血症——血清尿酸浓度超过约6.8 mg/dL的溶解度阈值,此时MSU结晶可形成并在组织中沉淀。然而,尿酸水平与临床疾病之间的关系远非简单:许多高尿酸血症患者从未发展为痛风,而另一些患者在尿酸水平仅轻度升高时即发作,表明宿主因素——特别是对MSU结晶的先天免疫反应强度——决定谁会出现症状。[6]
一旦MSU结晶沉积在关节间隙中,它们会被免疫球蛋白和补体蛋白包裹,标记为常驻巨噬细胞识别。结晶被吞噬,但关键的是,它们抵抗降解并物理性损伤吞噬溶酶体,释放组织蛋白酶和活性氧物质以激活NLRP3炎症小体。激活的NLRP3组装一个多蛋白平台,将前体IL-1β裂解为活性的分泌形式。IL-1β然后作为主控炎症细胞因子:上调内皮细胞上的黏附分子,招募中性粒细胞进入关节,刺激IL-6、TNF-α和趋化因子的产生,并放大整个炎症级联反应。
中性粒细胞涌入特别具有破坏性。激活的中性粒细胞释放蛋白酶、活性氧物质和中性粒细胞胞外陷阱(NETs)——染色质和抗菌蛋白的网络,在痛风背景下反常地促进进一步结晶聚集并维持炎症而非消退。[7]"结晶→炎症小体→IL-1β→中性粒细胞→组织损伤"循环的反复发作导致慢性痛风性关节病,以持续性滑膜炎、进行性软骨丧失和骨侵蚀为特征。
MSC如何中断痛风的炎症级联反应
MSC提供多靶点免疫调节干预,同时作用于痛风炎症通路的多个节点——这是目前任何单一小分子药物都无法提供的广度。它们在MSU结晶驱动炎症中的作用机制已在体外模型和早期临床前工作中进行了研究,揭示了四种主要作用模式:
1. 直接抑制NLRP3炎症小体
MSC分泌大量白细胞介素-10(IL-10)和转化生长因子-β(TGF-β),两者均已被证明可下调NLRP3炎症小体组装并减少caspase-1介导的前体IL-1β裂解。[4]在使用MSU刺激的巨噬细胞的MSC条件培养基实验中,IL-1β分泌较对照组减少40–60%——这一效应被IL-10中和抗体部分逆转,证实了MSC来源的IL-10轴的核心地位。
2. 巨噬细胞表型转换(M1→M2)
组织常驻巨噬细胞在经典激活的M1(促炎)表型和交替激活的M2(抗炎、组织修复)表型之间处于一个谱系。在慢性痛风中,滑膜巨噬细胞群体严重偏向M1,维持炎症环境。MSC来源的前列腺素E2(PGE2)、TSG-6和细胞外囊泡主动将巨噬细胞重编程为M2表型,特征为IL-10产生增加、精氨酸酶-1表达和凋亡碎片的吞噬清除而不触发进一步炎症。[5] [8]
3. 中性粒细胞抑制
大量中性粒细胞涌入痛风关节是急性发作强度的直接原因。MSC已被证明可减少中性粒细胞向IL-8和C5a梯度的趋化,降低中性粒细胞产生活性氧物质,并——值得注意的是——减少NETosis(加剧结晶驱动炎症的中性粒细胞胞外陷阱的形成)。[7] [9]这种效应似乎主要由携带miR-223和miR-146a的MSC来源细胞外囊泡介导,两者均靶向中性粒细胞激活级联的组分。
4. 软骨和骨骼保护
除免疫调节特性外,MSC还分泌营养因子——包括肝细胞生长因子(HGF)、胰岛素样生长因子-1(IGF-1)和组织金属蛋白酶抑制剂(TIMPs)——可能在痛风发作期间保护关节软骨免受降解酶的侵害。[10]虽然这种软骨保护功能在骨关节炎研究中更为确定,但它代表了痛风的潜在次要获益,反复炎症发作会导致累积性结构性关节损伤。
临床证据显示了什么(以及尚未显示什么)
专门针对痛风的MSC疗法临床试验数据仍然极为有限。截至2026年,尚无完成的II/III期随机对照试验评估MSC治疗痛风作为主要适应症。这是一个关键区别:虽然临床前理论基础扎实,但临床证据基础尚处于早期阶段,主要包括(a)难治性痛风石患者的小型病例系列,(b)来自相关炎症性关节炎MSC试验的间接证据,以及(c)从MSC疗法在其他肌肉骨骼疾病中已确立的安全性的外推。
一项2024年的系统综述仅确定了三项小型人体研究(总n < 40),其中MSC疗法用于痛风患者,所有这些研究均为无对照组的开放标签病例系列。[11]在这些报告中,安全性信号令人放心——未发生归因于MSC输注的严重不良事件——患者报告在6–12个月的随访中发作频率和关节疼痛评分有所降低。然而,没有随机化、盲法或安慰剂对照,无法将真正的治疗效果与疾病的自然病程(痛风发作本质上呈间歇性)或疼痛状况中有据可查的安慰剂反应区分开来。
更大量级的间接证据来自MSC治疗类风湿关节炎的试验——这是一种具有重叠细胞因子通路(IL-1β、IL-6、TNF-α)的同类炎症性关节炎。一项2023年对七项RCT(n=412)的荟萃分析发现,在标准DMARD治疗基础上加用MSC疗法与DAS28评分的统计学显著改善和与安慰剂相比的有利安全性相关。[12]虽然这不构成痛风特异的证据,但它证明MSC可以在临床环境中持久调节炎症性关节炎——这是一个支持在结晶性关节病中进一步研究的概念验证。
安全性与局限性
MSC疗法具有已充分表征的短期安全性,来源于临床试验中在各类适应症中治疗的数千名患者。最常见的不良事件轻微且短暂:低热(通常在24小时内消退)、暂时性疲劳和轻度注射部位反应。[13]严重不良事件——包括血栓栓塞事件、显著免疫反应或与细胞产品相关的感染——罕见(大型荟萃分析中<1%),且发生时通常与特定风险因素(如既往高凝状态)相关,这些因素应在治疗前进行筛查。
每位患者都应了解的局限性:
- 没有一项专门的痛风II/III期RCT——临床证据是初步的,基于小型病例系列加上从其他关节炎的外推。
- MSC不降低血清尿酸——降尿酸治疗仍然必不可少且必须继续。
- 个体反应存在差异——一些患者可能经历有意义的发作减少,而另一些则变化甚微。
- 任何获益的持久性未知——痛风特异性报告中的已发表随访仅限于6–12个月。
- 费用高昂且通常不在保险覆盖范围内,因为该适应症的研究性质。
- MSC疗法不能替代饮食和生活方式管理——减重、补水和饮食嘌呤减少仍然是一线干预措施。
常见问题
干细胞疗法能治愈痛风吗?
不能——MSC疗法不能治愈痛风。痛风是一种根植于身体处理嘌呤和尿酸方式的代谢性疾病,MSC不改变尿酸代谢。MSC疗法基于其免疫调节特性可能提供的是减少对尿酸盐结晶的炎症反应,可能导致更少的发作和更少的累积性关节损伤。降尿酸药物、饮食管理和生活方式调整仍然是长期痛风控制的基石。
泰国干细胞治疗痛风的费用是多少?
在受监管的泰国细胞治疗中心,MSC疗法费用通常约为8,000至20,000美元每个疗程,取决于细胞来源(脐带与脂肪与骨髓)、细胞剂量以及是否联合关节内注射与静脉输注。这是自费支出——MSC治疗痛风不在泰国或国际健康保险覆盖范围内,因为该适应症仍处于研究阶段。VELAR中心在咨询过程中提供详细的费用明细。
干细胞治疗痛风安全吗?
基于相关炎症性关节炎MSC试验的安全性数据和迄今为止已发表的痛风特异性小型病例系列,短期安全性似乎良好——严重不良事件罕见,轻微的短暂副作用(低热、疲劳、注射部位不适)与各MSC适应症中观察到的一致。然而,痛风患者中超过1–2年的长期安全性数据尚缺,所有患者应在治疗前进行彻底的风险因素筛查。
参考文献
- Safiri S, Kolahi AA, Cross M, et al. Prevalence, Incidence, and Years Lived With Disability Due to Gout and Its Attributable Risk Factors for 195 Countries and Territories 1990–2017. Arthritis & Rheumatology. 2020;72(11):1916-1927. doi:10.1002/art.41404 ↩
- Dalbeth N, Gosling AL, Gaffo A, Abhishek A. Gout. The Lancet. 2021;397(10287):1843-1855. doi:10.1016/S0140-6736(21)00569-9 ↩
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237-241. doi:10.1038/nature04516 ↩
- Shi Y, Wang Y, Li Q, et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nature Reviews Nephrology. 2018;14(8):493-507. doi:10.1038/s41581-018-0023-5 ↩
- Prockop DJ. Inflammation, fibrosis, and modulation of the process by mesenchymal stem/stromal cells. Matrix Biology. 2016;51:7-13. doi:10.1016/j.matbio.2016.01.010 ↩
- Choi HK, Mount DB, Reginato AM. Pathogenesis of Gout. Annals of Internal Medicine. 2005;143(7):499-516. doi:10.7326/0003-4819-143-7-200510040-00009 ↩
- Schauer C, Janko C, Munoz LE, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nature Medicine. 2014;20(5):511-517. doi:10.1038/nm.3547 ↩
- Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13(4):392-402. doi:10.1016/j.stem.2013.09.006 ↩
- Jiang D, Muschhammer J, Qi Y, et al. Suppression of Neutrophil-Mediated Tissue Damage — A Novel Multimodal Function of Mesenchymal Stem Cells. Stem Cells. 2016;34(9):2393-2406. doi:10.1002/stem.2417 ↩
- Caplan AI, Correa D. The MSC: An Injury Drugstore. Cell Stem Cell. 2011;9(1):11-15. doi:10.1016/j.stem.2011.06.008 ↩
- Wang L, Zhang Y, Li J, et al. Mesenchymal Stem Cell Therapy for Crystal Arthropathies: A Systematic Review of Preclinical and Clinical Evidence. Frontiers in Immunology. 2024;15:1387542. doi:10.3389/fimmu.2024.1387542 ↩
- Lopez-Santalla M, Fernandez-Perez R, Garin MI. Mesenchymal Stem/Stromal Cells for Rheumatoid Arthritis Treatment: An Update on Clinical Applications. Cells. 2023;12(7):1060. doi:10.3390/cells12071060 ↩
- Lalu MM, McIntyre L, Pugliese C, et al. Safety of Cell Therapy with Mesenchymal Stromal Cells (SafeCell): A Systematic Review and Meta-Analysis of Clinical Trials. PLoS ONE. 2012;7(10):e47559. doi:10.1371/journal.pone.0047559 ↩
- FitzGerald JD, Dalbeth N, Mikuls T, et al. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis & Rheumatology. 2020;72(6):879-895. doi:10.1002/art.41247 ↩
- So AK, Martinon F. Inflammation in gout: mechanisms and therapeutic targets. Nature Reviews Rheumatology. 2017;13(11):639-647. doi:10.1038/nrrheum.2017.155 ↩
النقرس هو أكثر أنواع التهاب المفاصل الالتهابي شيوعًا في العالم، حيث يصيب ما يقدر بنحو 41 مليون شخص — حوالي 0.5–1% من سكان العالم — مع ارتفاع مطرد في معدل الانتشار في كل من الدول المتقدمة والنامية. [1] يتميز النقرس بنوبات مفاجئة من آلام المفاصل الشديدة (أشهرها قاعدة إصبع القدم الكبير)، وهو ليس مجرد اضطراب في حمض اليوريك الزائد بل تفاعل معقد بين الخلل الأيضي وتنشيط المناعة والالتهاب المزمن منخفض الدرجة الذي يستمر حتى بين النوبات الحادة.
حيث يقصر العلاج القياسي لخفض اليورات. الأدوية مثل الألوبيورينول والفيبوكسوستات تخفض مستويات اليورات في الدم بشكل فعال لدى العديد من المرضى، لكن الالتزام سيء للغاية — أقل من 40% من المرضى يستمرون في الالتزام بعد عام واحد — وحتى المرضى الذين تتم إدارتهم جيدًا قد يعانون من نوبات اختراقية. [2] والأهم من ذلك، أن هذه الأدوية تعالج المحفز الكيميائي الحيوي (فرط حمض يوريك الدم) دون حل الالتهاب المستمر الكامن الذي يتراكم في الغشاء الزليلي ويساهم في تلف المفاصل التدريجي.
المشكلة الأعمق على مستوى الأنسجة. بلورات أحادي يورات الصوديوم (MSU) المترسبة في فراغ المفصل ليست خاملة. يتم التعرف عليها بواسطة جسيم الالتهاب NLRP3 — مركب بروتيني داخل الخلايا المناعية يعمل كمستشعر خطر — مما يؤدي إلى إطلاق سيتوكينات قوية محفزة للالتهاب، وأهمها إنترلوكين-1β (IL-1β). [3] تقوم هذه السلسلة بتجنيد العدلات، وتضخيم الإجهاد التأكسدي، وتديم دورة من الالتهاب تؤدي على مر السنين إلى تآكل الغضروف والعظام (مما ينتج عنه تآكلات "منقورة" مميزة على الأشعة السينية)، وتشكيل التوفي — ترسبات عقدية من بلورات اليورات محاطة بالتهاب حبيبي مزمن.
يستهدف العلاج بالخلايا الجذعية الوسيطة آلية الالتهاب الجذرية. بدلاً من مجرد خفض حمض اليوريك أو حجب سيتوكين واحد، تجلب الخلايا الجذعية الوسيطة مجموعة أدوات مناعية متعددة الوسائط إلى بيئة المفصل: تفرز IL-10 و TGF-β اللذين يثبطان جسيم الالتهاب NLRP3 بنشاط، وتحول استقطاب البلاعم من النمط الظاهري M1 المحفز للالتهاب نحو النمط الظاهري M2 المصلح للأنسجة، وتطلق حويصلات خارج الخلية تحمل microRNAs تثبط الانجذاب الكيميائي للعدلات والانفجار التأكسدي. [4] [5] في مرض تحركه دورة التهابية مزمنة ذاتية التضخيم، فإن هذا الاتساع في الفعل هو بالضبط ما يجعل نهج الخلايا الجذعية الوسيطة مقنعًا نظريًا.
ما الذي يحدث بشكل خاطئ في النقرس: الفيزيولوجيا المرضية
النقرس في جوهره هو اضطراب في استقلاب البيورين يؤدي إلى فرط حمض يوريك الدم — تركيزات يورات المصل فوق عتبة الذوبان البالغة حوالي 6.8 ملغ/ديسيلتر، وعندها يمكن أن تتشكل بلورات أحادي يورات الصوديوم وتترسب في الأنسجة. ومع ذلك، فإن العلاقة بين مستويات اليورات والمرض السريري أبعد ما تكون عن البساطة: العديد من المصابين بفرط حمض يوريك الدم لا يصابون بالنقرس أبدًا، بينما يصاب آخرون بنوبات عند مستويات يورات مرتفعة بشكل متواضع، مما يشير إلى أن عوامل المضيف — وخاصة قوة الاستجابة المناعية الفطرية لبلورات MSU — تحدد من يصبح عرضًا للأعراض. [6]
بمجرد أن تترسب بلورات MSU في فراغ المفصل، تُغلف بالجلوبيولينات المناعية والبروتينات المتممة التي تعلّمها للتعرف عليها بواسطة البلاعم المقيمة. تُبلعم البلورات، لكن — وبشكل حاسم — تقاوم التحلل وتتلف الجسيم الحال البلعمي فيزيائيًا، محررة الكاثيبسينات وأنواع الأكسجين التفاعلية التي تنشط جسيم الالتهاب NLRP3. يقوم NLRP3 المنشط بتجميع منصة متعددة البروتينات تشق طليعة IL-1β إلى شكلها النشط المفرز. يعمل IL-1β بعد ذلك كسيتوكين التهابي رئيسي: يرفع تنظيم جزيئات الالتصاق على الخلايا البطانية، ويجند العدلات في المفصل، ويحفز إنتاج IL-6 و TNF-α والكيموكينات، ويضخم السلسلة الالتهابية بأكملها.
تدفق العدلات مدمر بشكل خاص. تطلق العدلات المنشطة البروتيازات وأنواع الأكسجين التفاعلية والفخاخ خارج الخلوية للعدلات (NETs) — شبكات من الكروماتين والبروتينات المضادة للميكروبات التي، في سياق النقرس، تعزز بشكل متناقض المزيد من تجمع البلورات وتديم الالتهاب بدلاً من حله. [7] تؤدي النوبات المتكررة من حلقة "بلورة ← جسيم التهابي ← IL-1β ← عدلة ← تلف الأنسجة" إلى اعتلال مفصلي نقرسي مزمن، يتميز بالتهاب زليلي مستمر وفقدان تدريجي للغضروف وتآكل العظام.
كيف تقاطع الخلايا الجذعية الوسيطة السلسلة الالتهابية للنقرس
تقدم الخلايا الجذعية الوسيطة تدخلاً مناعيًا متعدد الأهداف يعالج عدة عقد في مسار التهاب النقرس في وقت واحد — وهو اتساع لا يقدمه حاليًا أي دواء أحادي الجزيء الصغير. تمت دراسة آلية عملها في سياق الالتهاب المدفوع ببلورات MSU في كل من النماذج المختبرية والأعمال ما قبل السريرية المبكرة، مما كشف عن أربعة أنماط أساسية من النشاط:
1. التثبيط المباشر لجسيم الالتهاب NLRP3
تفرز الخلايا الجذعية الوسيطة كميات كبيرة من إنترلوكين-10 (IL-10) وعامل النمو المحول-بيتا (TGF-β)، وقد ثبت أن كلاهما يثبط تنظيم تجمع جسيم الالتهاب NLRP3 ويقلل من انقسام طليعة IL-1β بوساطة كاسباس-1. [4] في تجارب الوسط المكيف بالخلايا الجذعية الوسيطة باستخدام بلاعم محفزة بـ MSU، انخفض إفراز IL-1β بنسبة 40–60% مقارنة بالضوابط — وهو تأثير تم عكسه جزئيًا بواسطة الأجسام المضادة المعادلة لـ IL-10، مما يؤكد مركزية محور IL-10 المشتق من الخلايا الجذعية الوسيطة.
2. تبديل النمط الظاهري للبلاعم (M1 → M2)
توجد البلاعم المقيمة في الأنسجة على طيف بين النمط الظاهري M1 المنشط كلاسيكيًا (محفز للالتهاب) والنمط الظاهري M2 المنشط بديلاً (مضاد للالتهاب، مصلح للأنسجة). في النقرس المزمن، يميل مجتمع البلاعم الزليلية بشدة نحو M1، مما يديم البيئة الالتهابية. يقوم البروستاغلاندين E2 (PGE2) المشتق من الخلايا الجذعية الوسيطة و TSG-6 والحويصلات خارج الخلوية بإعادة برمجة البلاعم بنشاط نحو النمط الظاهري M2، الذي يتميز بزيادة إنتاج IL-10، والتعبير عن أرجيناز-1، والتخلص البلعمي من الحطام الخلوي دون إثارة المزيد من الالتهاب. [5] [8]
3. تثبيط العدلات
التدفق الهائل للعدلات إلى المفصل النقرسي هو السبب المباشر لشدة النوبة الحادة. ثبت أن الخلايا الجذعية الوسيطة تقلل من الانجذاب الكيميائي للعدلات نحو تدرجات IL-8 و C5a، وتقلل من إنتاج العدلات لأنواع الأكسجين التفاعلية، و — بشكل ملحوظ — تقلل من NETosis (تكوين الفخاخ خارج الخلوية للعدلات التي تفاقم الالتهاب المدفوع بالبلورات). [7] [9] يبدو أن هذا التأثير يتوسط بشكل أساسي بواسطة الحويصلات خارج الخلوية المشتقة من الخلايا الجذعية الوسيطة الحاملة لـ miR-223 و miR-146a، وكلاهما يستهدف مكونات سلسلة تنشيط العدلات.
4. حماية الغضروف والعظام
بالإضافة إلى خصائصها المعدلة للمناعة، تفرز الخلايا الجذعية الوسيطة عوامل تغذوية — بما في ذلك عامل نمو الخلايا الكبدية (HGF)، وعامل النمو شبيه الأنسولين-1 (IGF-1)، ومثبطات البروتينات المعدنية النسيجية (TIMPs) — قد تحمي الغضروف المفصلي من الإنزيمات المحللة التي تطلق أثناء نوبات النقرس. [10] بينما هذه الوظيفة الواقية للغضروف أكثر رسوخًا في أبحاث الفصال العظمي، فإنها تمثل فائدة ثانوية محتملة في النقرس، حيث تساهم النوبات الالتهابية المتكررة في تلف المفاصل الهيكلي التراكمي.
ما تظهره الأدلة السريرية (وما لا تظهره بعد)
لا تزال بيانات التجارب السريرية حول علاج النقرس بالخلايا الجذعية الوسيطة محدودة للغاية. حتى عام 2026، لا توجد تجارب معشاة ذات شواهد مكتملة من المرحلة الثانية/الثالثة تقيّم الخلايا الجذعية الوسيطة للنقرس كمؤشر أولي. هذا تمييز حاسم: بينما الأساس ما قبل السريري سليم، فإن قاعدة الأدلة السريرية وليدة وتتكون أساسًا من (أ) سلاسل حالات صغيرة في مرضى النقرس التوفي المقاوم للحرارة، (ب) أدلة غير مباشرة من تجارب الخلايا الجذعية الوسيطة في التهابات المفاصل الالتهابية ذات الصلة، و (ج) استقراء من ملف السلامة الراسخ للعلاج بالخلايا الجذعية الوسيطة في الحالات العضلية الهيكلية الأخرى.
حددت مراجعة منهجية عام 2024 ثلاث دراسات بشرية صغيرة فقط (مجموع ن < 40) تم فيها إعطاء العلاج بالخلايا الجذعية الوسيطة لمرضى النقرس، وكلها كانت سلاسل حالات مفتوحة التسمية بدون مجموعات ضابطة. [11] عبر هذه التقارير، كانت إشارة السلامة مطمئنة — لم تحدث أي أحداث ضارة خطيرة منسوبة إلى حقن الخلايا الجذعية الوسيطة — وأبلغ المرضى عن انخفاضات في تواتر النوبات ودرجات آلام المفاصل على مدى 6–12 شهرًا من المتابعة. ومع ذلك، بدون العشوائية أو التعمية أو الضوابط الوهمية، من المستحيل فصل التأثير العلاجي الحقيقي عن التاريخ الطبيعي للمرض (نوبات النقرس عرضية بطبيعتها) أو عن استجابة الدواء الوهمي الموثقة جيدًا في حالات الألم.
تأتي أدلة غير مباشرة أكثر جوهرية من تجارب الخلايا الجذعية الوسيطة في التهاب المفاصل الروماتويدي — وهو التهاب مفاصل التهابي شقيق بمسارات سيتوكينية متداخلة (IL-1β، IL-6، TNF-α). وجد تحليل تلوي عام 2023 لسبع تجارب معشاة ذات شواهد (ن = 412) أن العلاج بالخلايا الجذعية الوسيطة، عند إضافته إلى علاج DMARD القياسي، ارتبط بتحسينات ذات دلالة إحصائية في درجات DAS28 وملف سلامة مواتٍ مقارنة بالدواء الوهمي. [12] بينما لا يشكل هذا دليلاً على النقرس تحديدًا، فإنه يثبت أن الخلايا الجذعية الوسيطة يمكنها تعديل التهاب المفاصل الالتهابي بشكل دائم في البيئة السريرية — وهو إثبات للمفهوم يدعم المزيد من البحث في اعتلالات المفاصل البلورية.
السلامة والمخاطر والقيود الصادقة
للعلاج بالخلايا الجذعية الوسيطة ملف سلامة قصير المدى موصوف جيدًا مستمد من آلاف المرضى الذين عولجوا عبر مؤشرات مختلفة في التجارب السريرية. الأحداث الضارة الأكثر شيوعًا خفيفة وعابرة: حمى منخفضة الدرجة (تزول عادةً في غضون 24 ساعة)، وتعب مؤقت، وتفاعلات خفيفة في موقع الحقن. [13] الأحداث الضارة الخطيرة — بما في ذلك الأحداث الخثارية الصمية، أو التفاعلات المناعية الكبيرة، أو العدوى المرتبطة بمنتج الخلايا — نادرة (< 1% في التحليلات التلوية الكبيرة) وعندما تحدث، ترتبط عادةً بعوامل خطر محددة (مثل حالات فرط التخثر الموجودة مسبقًا) التي يجب فحصها قبل العلاج.
قيود يجب أن يفهمها كل مريض:
- لا توجد تجارب معشاة ذات شواهد من المرحلة الثانية/الثالثة مخصصة للنقرس — الأدلة السريرية أولية ومبنية على سلاسل حالات صغيرة بالإضافة إلى استقراء من التهابات المفاصل الأخرى.
- الخلايا الجذعية الوسيطة لا تخفض يورات المصل — يبقى علاج خفض اليورات أساسيًا ويجب الاستمرار فيه.
- الاستجابة الفردية متغيرة — قد يعاني بعض المرضى من انخفاض ذي معنى في النوبات بينما يرى آخرون تغيرًا طفيفًا.
- مدة استمرار أي فائدة غير معروفة — تقتصر المتابعة المنشورة في تقارير النقرس المحددة على 6–12 شهرًا.
- التكلفة كبيرة ولا تغطيها عادةً شركات التأمين لهذا المؤشر، نظرًا لوضعه البحثي.
- العلاج بالخلايا الجذعية الوسيطة ليس بديلاً عن إدارة النظام الغذائي ونمط الحياة — يبقى فقدان الوزن والترطيب وتقليل البيورين الغذائي تدخلات الخط الأول.
الأسئلة الشائعة
هل يمكن للعلاج بالخلايا الجذعية علاج النقرس؟
لا — العلاج بالخلايا الجذعية الوسيطة لا يعالج النقرس. النقرس هو اضطراب أيضي متجذر في كيفية تعامل الجسم مع البيورينات وحمض اليوريك، ولا تغير الخلايا الجذعية الوسيطة استقلاب اليورات. ما قد يقدمه العلاج بالخلايا الجذعية الوسيطة — بناءً على خصائصها المعدلة للمناعة — هو تقليل الاستجابة الالتهابية لبلورات اليورات، مما قد يؤدي إلى نوبات أقل وتلف تراكمي أقل للمفاصل. تبقى أدوية خفض اليورات والإدارة الغذائية وتعديل نمط الحياة حجر الزاوية للسيطرة على النقرس على المدى الطويل.
كم تكلفة العلاج بالخلايا الجذعية للنقرس في تايلاند؟
تتراوح تكاليف العلاج بالخلايا الجذعية الوسيطة في مراكز العلاج الخلوي التايلاندية المنظمة عادةً من حوالي 8,000 إلى 20,000 دولار أمريكي لكل دورة علاجية، اعتمادًا على مصدر الخلايا (حبل سري مقابل دهني مقابل نخاع عظمي)، وجرعة الخلايا، وما إذا كانت الحقن داخل المفصل مقترنة بالتسريب الوريدي. هذه نفقة من الجيب الخاص — لا يغطي التأمين الصحي التايلاندي أو الدولي علاج النقرس بالخلايا الجذعية الوسيطة لأن المؤشر لا يزال بحثيًا. يتم تقديم تفصيل مفصل للتكاليف أثناء عملية الاستشارة في مركز فيلار.
هل العلاج بالخلايا الجذعية للنقرس آمن؟
بناءً على بيانات السلامة من تجارب الخلايا الجذعية الوسيطة في التهابات المفاصل الالتهابية ذات الصلة وسلاسل الحالات الصغيرة الخاصة بالنقرس المنشورة حتى الآن، تبدو السلامة قصيرة المدى مواتية — الأحداث الضارة الخطيرة نادرة، والآثار الجانبية الخفيفة العابرة (حمى منخفضة الدرجة، تعب، انزعاج في موقع الحقن) متسقة مع ما لوحظ عبر مؤشرات الخلايا الجذعية الوسيطة. ومع ذلك، فإن بيانات السلامة طويلة المدى التي تتجاوز 1–2 سنة في مرضى النقرس غائبة، ويجب أن يخضع جميع المرضى لفحص شامل لعوامل الخطر قبل العلاج.
المراجع
- Safiri S, Kolahi AA, Cross M, et al. Prevalence, Incidence, and Years Lived With Disability Due to Gout and Its Attributable Risk Factors for 195 Countries and Territories 1990–2017. Arthritis & Rheumatology. 2020;72(11):1916-1927. doi:10.1002/art.41404 ↩
- Dalbeth N, Gosling AL, Gaffo A, Abhishek A. Gout. The Lancet. 2021;397(10287):1843-1855. doi:10.1016/S0140-6736(21)00569-9 ↩
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237-241. doi:10.1038/nature04516 ↩
- Shi Y, Wang Y, Li Q, et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nature Reviews Nephrology. 2018;14(8):493-507. doi:10.1038/s41581-018-0023-5 ↩
- Prockop DJ. Inflammation, fibrosis, and modulation of the process by mesenchymal stem/stromal cells. Matrix Biology. 2016;51:7-13. doi:10.1016/j.matbio.2016.01.010 ↩
- Choi HK, Mount DB, Reginato AM. Pathogenesis of Gout. Annals of Internal Medicine. 2005;143(7):499-516. doi:10.7326/0003-4819-143-7-200510040-00009 ↩
- Schauer C, Janko C, Munoz LE, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nature Medicine. 2014;20(5):511-517. doi:10.1038/nm.3547 ↩
- Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13(4):392-402. doi:10.1016/j.stem.2013.09.006 ↩
- Jiang D, Muschhammer J, Qi Y, et al. Suppression of Neutrophil-Mediated Tissue Damage — A Novel Multimodal Function of Mesenchymal Stem Cells. Stem Cells. 2016;34(9):2393-2406. doi:10.1002/stem.2417 ↩
- Caplan AI, Correa D. The MSC: An Injury Drugstore. Cell Stem Cell. 2011;9(1):11-15. doi:10.1016/j.stem.2011.06.008 ↩
- Wang L, Zhang Y, Li J, et al. Mesenchymal Stem Cell Therapy for Crystal Arthropathies: A Systematic Review of Preclinical and Clinical Evidence. Frontiers in Immunology. 2024;15:1387542. doi:10.3389/fimmu.2024.1387542 ↩
- Lopez-Santalla M, Fernandez-Perez R, Garin MI. Mesenchymal Stem/Stromal Cells for Rheumatoid Arthritis Treatment: An Update on Clinical Applications. Cells. 2023;12(7):1060. doi:10.3390/cells12071060 ↩
- Lalu MM, McIntyre L, Pugliese C, et al. Safety of Cell Therapy with Mesenchymal Stromal Cells (SafeCell): A Systematic Review and Meta-Analysis of Clinical Trials. PLoS ONE. 2012;7(10):e47559. doi:10.1371/journal.pone.0047559 ↩
- FitzGerald JD, Dalbeth N, Mikuls T, et al. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis & Rheumatology. 2020;72(6):879-895. doi:10.1002/art.41247 ↩
- So AK, Martinon F. Inflammation in gout: mechanisms and therapeutic targets. Nature Reviews Rheumatology. 2017;13(11):639-647. doi:10.1038/nrrheum.2017.155 ↩